
ELP1&ELP1 IVS20+6T>C家族性自主神經功能障礙人源化模型的介紹
在基因轉錄過程中,外顯子和內含子都會被復制到前體信使RNA(pre-mRNA)中,隨后通過剪接去除內含子,獲得編碼序列,最終翻譯成蛋白質。隨著全基因組測序的發展,人們逐漸認識到內含子突變在多種疾病,尤其是罕見病中,具有重要作用。研究表明,約10%-30%的疾病相關基因突變(主要為內含子突變)會影響剪接或通過調控元件(如增強子和沉默子)的失調,引發隱性剪接位點激活、假外顯子包含和外顯子跳躍等機制,最終導致疾病。這些機制通常會產生提前終止密碼子(PTC),引發無義介導的RNA降解(NMD)、蛋白質二級結構改變或基因/蛋白表達水平失調[1-3]。今天為大家介紹的家族性自主神經功能障礙研究模型,即是由內含子突變引發的典型疾病案例。

圖1 內含子突變對Pre-mRNA剪接的常見影響類型[3]
家族性自主神經功能障礙(FD)與ELP基因內含子突變
家族性自主神經功能障礙(FD)是一種罕見的遺傳性神經系統疾病,由神經元發育受損和中樞神經系統退化引起。患者因自主神經系統和感覺神經系統的缺陷,表現出多汗、間歇性高血壓、流涎、吞咽困難、大小便失常、呼吸困難及周期性嘔吐等癥狀。該病具有種族特異性,主要影響德系猶太人,在該人群中的發病率約為1/3600,僅約50%的患者能存活至40歲[4-5]。ELP1(IKBKAP)基因編碼延伸復合物組成部分,在神經元的發育和功能中發揮重要作用。幾乎所有FD患者都攜帶ELP1基因雙拷貝突變,其中超過99%為第20號內含子5'剪接位點突變(IVS20+6T>C)。該突變破壞了U1小核核糖核蛋白與第20號內含子供體剪接位點的堿基配對,導致第20號外顯子跳躍[4-6]。這種錯誤剪接導致轉錄本框架移位,產生提前終止密碼子(PTC),從而翻譯出截短的ELP蛋白,最終導致神經元損傷和死亡。
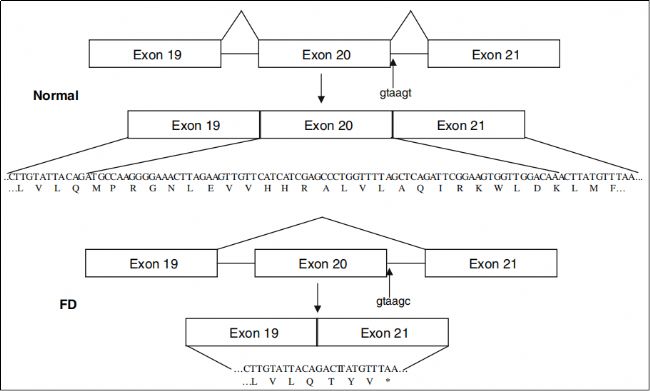
圖2 ELP1/IKBKAP內含子突變IVS20+6T>C導致轉錄本剪接異常的機制[6]
靶向ELP1的FD療法及相關動物模型
目前,FD尚無根治方法,治療策略主要集中在對癥治療和支持性護理,以緩解癥狀并預防并發癥。由于IVS20+6T>C突變是FD最常見的致病突變,研究的重點在于糾正該突變引起的錯誤剪接模式,以生成全長ELP1蛋白。冷泉港實驗室和PTC Therapeutics的研究人員在這一領域開展了諸多研究,包括反義寡核苷酸(ASO)和小分子藥物的開發[7-9]。研究表明,使用野生型小鼠研究內含子突變ELP1基因的剪接模式不可行,而純合Elp1基因敲除小鼠會在胚胎期死亡。表達帶有人源突變ELP1基因的轉基因小鼠由于正常水平的內源性小鼠Elp1基因表達,未表現出明顯的疾病表型,因此需要結合小鼠內源性Elp1基因單拷貝敲除,但這仍存在轉基因拷貝不穩定和表型不一致等問題[10-12]。此外,由于小鼠和人類基因剪接模式的差異,將小鼠Elp1基因第20號外顯子及其兩側內含子人源化并引入IVS20+6T>C突變,同樣未能產生表型[13]。
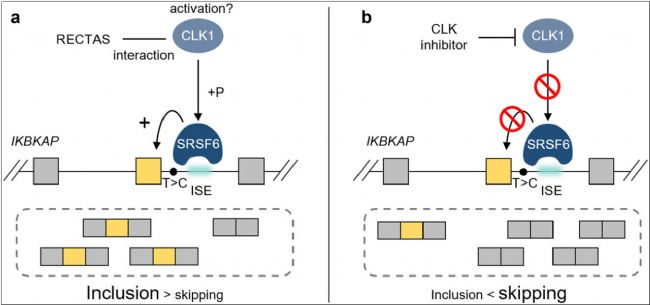
圖3 通過小分子藥物調控IKBKAP IVS20+6T>C第20號外顯子剪接模式[9]
以上研究表明,在小鼠體內研究ELP1基因剪接模式可能需要更長甚至全長人類ELP1基因序列。針對這一需求,賽業生物研發了小鼠Elp1基因人源化的B6-hELP1模型(產品編號:I001203),其中小鼠Elp1基因從起始密碼子到終止密碼子的序列被原位替換為人源ELP1基因的對應序列。此外,在此基礎上還構建了IVS20+6T>C人源化疾病模型,以滿足廣大科研人員在FD研究中的需求。
B6-hELP1小鼠成功表達人源ELP1基因
檢測結果表明,在B6-hELP1小鼠的大腦皮層、腎臟、肝臟、骨骼肌和心臟中均存在人源ELP1基因的顯著表達,且不存在鼠源Elp1基因的表達。
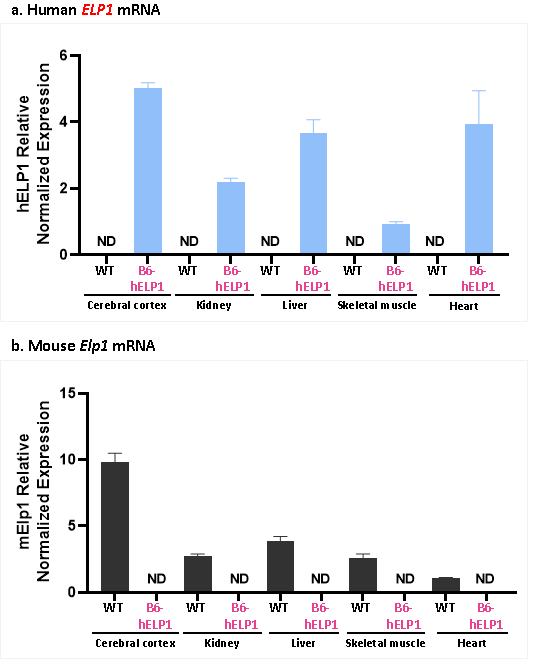
圖4 B6-hELP1小鼠和野生型小鼠體內人源ELP1基因和鼠源Elp1基因的表達
總 結
目前FD治療研究主要集中在糾正突變引起的錯誤剪接模式,以生成全長ELP1蛋白。B6-hELP1模型(產品編號:I001203)在小鼠體內表達全長的人源ELP1基因,并且不存在小鼠內源性Elp1基因的干擾,可用于FD研究。根據前期研究,基于B6-hELP1小鼠構建的B6-hELP1 IVS20+6T>C人源化點突變模型(在研)預計將出現與人類FD相似的表型。
此外,賽業生物在神經、眼科等疾病研究領域開發了多種遺傳疾病模型和人源化模型,為研究人員開發針對不同疾病的靶向藥物提供了有力支持。
HUGO-GT®全基因組人源化模型
參考文獻:
[1]Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, Chow ED, Kanterakis E, Gao H, Kia A, Batzoglou S, Sanders SJ, Farh KK. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019 Jan 24;176(3):535-548.e24.
[2]Chiang HL, Chen YT, Su JY, Lin HN, Yu CA, Hung YJ, Wang YL, Huang YT, Lin CL. Mechanism and modeling of human disease-associated near-exon intronic variants that perturb RNA splicing. Nat Struct Mol Biol. 2022 Nov;29(11):1043-1055.
[3]Lord J, Baralle D. Splicing in the Diagnosis of Rare Disease: Advances and Challenges. Front Genet. 2021 Jul 1;12:689892.
[4]Rubin BY, Anderson SL. IKBKAP/ELP1 gene mutations: mechanisms of familial dysautonomia and gene-targeting therapies. Appl Clin Genet. 2017 Dec 15;10:95-103.
[5]Dietrich P, Dragatsis I. Familial Dysautonomia: Mechanisms and Models. Genet Mol Biol. 2016 Oct-Dec;39(4):497-514. doi: 10.1590/1678-4685-GMB-2015-0335. Epub 2016 Aug 4.
[6]Rubin BY, Anderson SL. The molecular basis of familial dysautonomia: overview, new discoveries and implications for directed therapies. Neuromolecular Med. 2008;10(3):148-56.
[7]Morini E, Gao D, Montgomery CM, Salani M, Mazzasette C, Krussig TA, Swain B, Dietrich P, Narasimhan J, Gabbeta V, Dakka A, Hedrick J, Zhao X, Weetall M, Naryshkin NA, Wojtkiewicz GG, Ko CP, Talkowski ME, Dragatsis I, Slaugenhaupt SA. ELP1 Splicing Correction Reverses Proprioceptive Sensory Loss in Familial Dysautonomia. Am J Hum Genet. 2019 Apr 4;104(4):638-650.
[8]Sinha R, Kim YJ, Nomakuchi T, Sahashi K, Hua Y, Rigo F, Bennett CF, Krainer AR. Antisense oligonucleotides correct the familial dysautonomia splicing defect in IKBKAP transgenic mice. Nucleic Acids Res. 2018 Jun 1;46(10):4833-4844.
[9]Ajiro M, Awaya T, Kim YJ, Iida K, Denawa M, Tanaka N, Kurosawa R, Matsushima S, Shibata S, Sakamoto T, Studer L, Krainer AR, Hagiwara M. Therapeutic manipulation of IKBKAP mis-splicing with a small molecule to cure familial dysautonomia. Nat Commun. 2021 Jul 23;12(1):4507.
[10]Dietrich P, Yue J, E S, Dragatsis I. Deletion of exon 20 of the Familial Dysautonomia gene Ikbkap in mice causes developmental delay, cardiovascular defects, and early embryonic lethality. PLoS One. 2011;6(10):e27015.
[11]Hims MM, Shetty RS, Pickel J, Mull J, Leyne M, Liu L, Gusella JF, Slaugenhaupt SA. A humanized IKBKAP transgenic mouse models a tissue-specific human splicing defect. Genomics. 2007 Sep;90(3):389-96.
[12]Morini E, Dietrich P, Salani M, Downs HM, Wojtkiewicz GR, Alli S, Brenner A, Nilbratt M, LeClair JW, Oaklander AL, Slaugenhaupt SA, Dragatsis I. Sensory and autonomic deficits in a new humanized mouse model of familial dysautonomia. Hum Mol Genet. 2016 Mar 15;25(6):1116-28.
[13]Bochner R, Ziv Y, Zeevi D, Donyo M, Abraham L, Ashery-Padan R, Ast G. Phosphatidylserine increases IKBKAP levels in a humanized knock-in IKBKAP mouse model. Hum Mol Genet. 2013 Jul 15;22(14):2785-94.



