
罕見病Leber先天性黑矇的致病機理介紹及相關動物模型的應用
Leber先天性黑矇(Leber Congenital Amaurosis, LCA)是一種極其罕見的遺傳性視網膜疾病,以其嚴重的致盲性特征而著稱。患者往往在年幼時便出現癥狀,視功能受到顯著損害。臺灣學者的一項研究揭示了LCA患者的發病年齡普遍偏早,并且他們常在青少年時期尋求醫療幫助[1]。早在1869年,Theodore Leber博士便首次描述了患有眼球震顫和瞳孔光反射異常的嬰兒所展現出的嚴重視力問題,這被認為是LCA的典型表征[2]。
此外,流行病學報道顯示LCA占據了全部遺傳性視網膜營養不良癥的5%,其患病率約為每81,000至30,000人中就有1人受到影響。此外,LCA還占據了學齡兒童失明原因的20%,顯示了其對兒童視力的嚴重威脅[1,3-5]。
表型特點與診斷標準
LCA的主要特征顯著,包括:在出生或出生后不久即展現出的嚴重視力喪失,常伴隨眼球震顫(不自主的眼球運動)、瞳孔反射遲鈍、畏光或夜盲等癥狀。其視網膜電圖(ERG)顯示,各波形記錄幾乎不存在或顯著降低。LCA通常在患兒出生后6個月內發病,家長往往因發現孩子眼球震顫、無法注視或斜視而尋求醫療幫助[6]。
除了上述癥狀外,LCA還有其他一些常見的臨床表現。如圖1所示,患者的眼底損傷表現為視網膜血管組織減少,視網膜變薄,黃斑區出現變化,且缺乏中央暗斑。此外,患者還可能面臨視力持續下降、屈光不正等問題[7]。這些特征為醫生提供了診斷和治療LCA的重要依據。
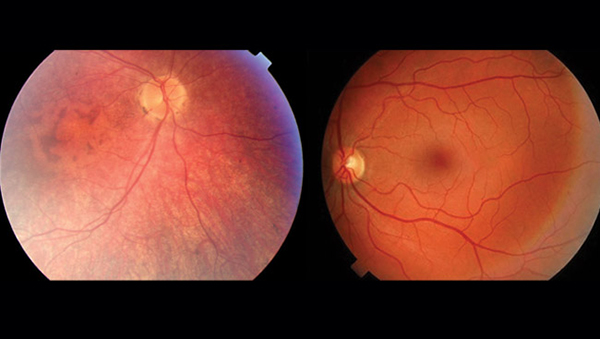
圖1 LCA患者與健康人眼底表現比較
(左為LCA患者,右為正常人)
關于LCA診斷標準[8],具體如下:
(1)患者6月齡前出現嚴重視力低下或盲,可伴有眼球震顫、指眼征、黑瞳孔等;
(2)ERG各波形記錄不到或嚴重降低;
(3)不伴有或伴有其他眼部或其他系統的先天發育異常。
致病基因與突變
根據臨床驗證和遺傳學分析,共有23個基因與LCA相關,其中最常見的基因為GUCY2D、RPE65、CRB1、CEP290等[9]。
GUCY2D
GUCY2D(圖2)是第一個被鑒定為與LCA相關的基因,位于染色體17p13.1上,編碼視網膜鳥苷酸環化酶-1,該酶參與光轉導中的感光器恢復階段。GUCY2D突變占LCA病例的6-21%。已知GUCY2D相關LCA病例在生命早期視力極差但靜止不動,伴有眼球震顫、眼指征和明顯的畏光[10,11]。
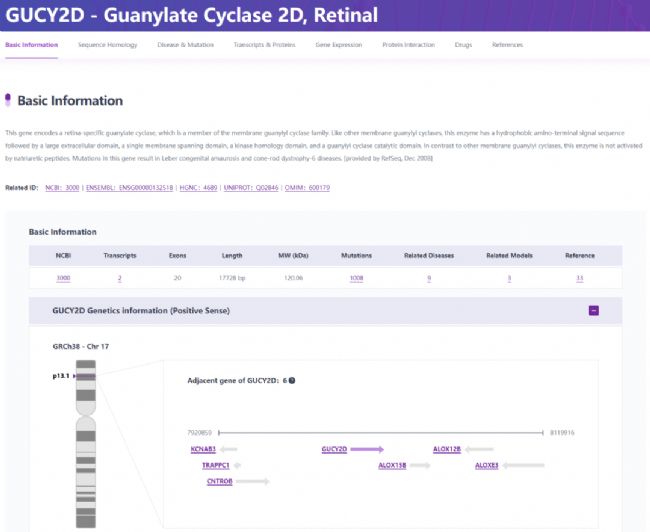
圖2 GUCY2D基因概況
RPE65
染色體1p31.3上的RPE65基因編碼類視黃醇異構酶(圖3),這是一種在RPE中大量表達的61kDa酶,負責類維生素A循環中的維生素A代謝。這種缺陷導致缺乏11-順式視網膜再生,從而影響視力。RPE65突變占LCA病例的4%-16%,在中國人群中相對罕見,但在高加索和印度人群中更為普遍[12]。
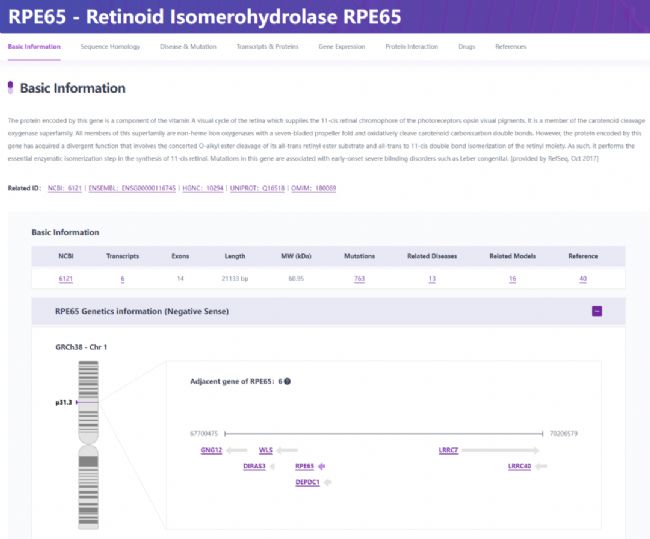
圖3 RPE65基因概況
(圖片來源:RDDC罕見病數據中心)
該基因與LCA2最為密切相關,根據RDDC數據庫的統計(圖4),RPE65基因目前已確認存在639個與LCA2相關的突變,其中127個被認定為致病性突變,這些突變已被廣泛研究和報道。

圖4 RPE65基因突變與LCA2
(圖片來源:RDDC罕見病數據中心)
疾病模型
動物模型的使用成為了不可或缺的工具。這些模型包括了我們熟知的斑馬魚、小鼠、大鼠、貓、犬以及鳥類等,它們各自獨特的生物學特性為LCA的研究提供了豐富的實驗平臺。
斑馬魚作為一種在遺傳學研究中廣泛應用的模式生物,其眼睛結構與人類具有高度的相似性,使得斑馬魚成為研究視網膜發育和疾病機制的理想模型。特別是在模擬LCA相關的視網膜病變方面,斑馬魚模型能夠精準地重現疾病過程,為研究疾病的發病機制提供了直觀的證據。
犬類模型在LCA研究中同樣占據了重要地位。相較于其他動物,犬類的壽命較長,這使得研究人員能夠長期觀察疾病的進展和演變,為評估治療效果提供了更廣闊的時間窗口。此外,犬類模型還允許研究人員在更接近自然環境下研究LCA,從而得到更為貼近實際的實驗結果。
在LCA研究中,小鼠模型更是發揮著舉足輕重的作用。由于小鼠在遺傳和生理上與人類具有高度相似性,因此小鼠模型能夠非常準確地模擬人類LCA患者的基因突變和病理特征。借助現代基因編輯技術,如CRISPR-Cas9,科學家們能夠精確地在小鼠身上模擬人類LCA患者的基因突變,并觀察這些突變如何影響視網膜的結構和功能。這些小鼠模型不僅展示了與人類LCA患者相似的病理特征,如光感受器細胞的逐漸退化、視覺功能的損失以及視網膜的病變,還為評估潛在的治療方法提供了實驗基礎。為了更深入地理解LCA的發病機制和開發潛在的治療方法,研究人員已經建立了多種小鼠模型來模擬這一疾病(圖5)。
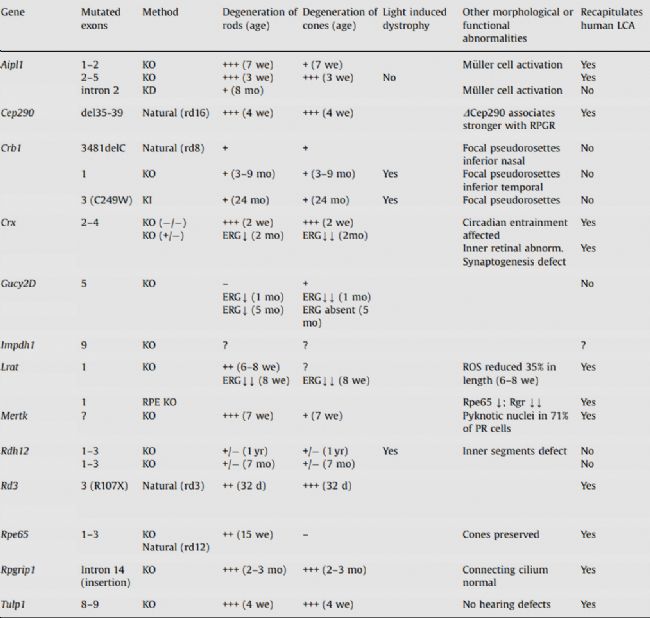
圖5 用于LCA的小鼠模型[13]
不同的小鼠模型各具特點:
Cep290敲除小鼠:這些小鼠缺乏CEP290基因,模擬了人類LCA患者中CEP290突變導致的視網膜病變。這些小鼠的視網膜發育不全,感光細胞逐漸退化,最終導致視力喪失;
Gucy2d敲除小鼠:這種小鼠模型模擬了GUCY2D基因突變導致的LCA。由于GUCY2D是光傳導過程中的關鍵酶,這些小鼠的感光細胞在光刺激下無法產生正常的信號,導致視力受損;
RPE65敲除小鼠由于缺乏或異常的RPE65酶活性,無法有效將全反式視黃醛轉化為11-順式視黃醛,導致感光細胞的功能受損。這種功能損害進而引發視網膜病變,包括視力下降、夜盲癥和視網膜色素變性等癥狀[13]。
通過這些小鼠模型,研究人員能夠更直接地觀察LCA的病理過程,并測試潛在的治療方法。這些模型為LCA的研究和治療提供了重要的工具。
治療方法
基因增強療法是LCA潛在治療的主要來源。已經對常見涉及的基因進行了動物研究,包括GUCY2D、RPE65、AIPL1、RPGRIP1、LCA5、CEP290和RDH12,主要是通過使用腺相關病毒(adeno-associated virus, AAV)載體介導或慢病毒載體介導的基因增強療法[6],載體的注射途徑如圖6所示,注射視網膜下或玻璃體內[14]。
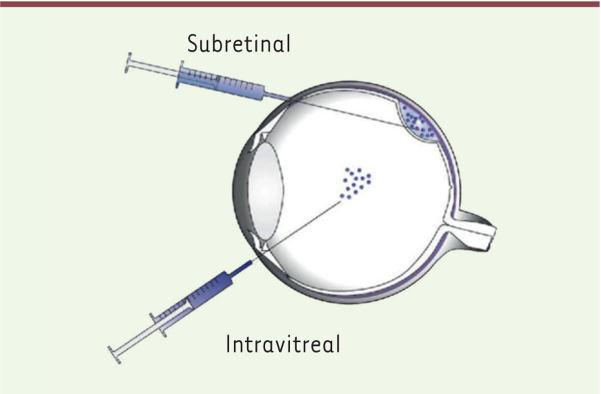
圖6 基因治療載體注射途徑[14]
特別值得一提的是,由RPE65基因突變引起的LCA2具有其獨特性。在此疾病中,視網膜神經感覺細胞在生命早期的保全尤為重要,因為基因治療的核心思想是通過引入功能性野生型RPE65基因來替換受損基因,進而維持相對完整的視覺功能[15]。
根據智慧芽新藥情報庫中的LCA基因治療藥物統計(圖7)可知:目前,針對RPE65突變的基因治療已取得顯著進展,其中Voreligene Neparvovec已進入上市階段。此外,針對GUCY2D、RPE65和CEP290的臨床試驗也正如火如荼地進行中,這些試驗旨在進一步驗證基因增強療法在治療LCA中的有效性和安全性。值得注意的是,多個國家都積極參與到LCA基因治療藥物的研發中。在中國,輝大(上海)生物科技有限公司和啟昇(上海)生物科技有限公司等公司也在該領域取得了重要進展。其中,輝大(上海)生物科技有限公司研發的藥物已進入臨床1/2期試驗,而啟昇(上海)生物科技有限公司的藥物則已進入臨床前階段,這些進展為LCA患者帶來了新的治療選擇和希望。
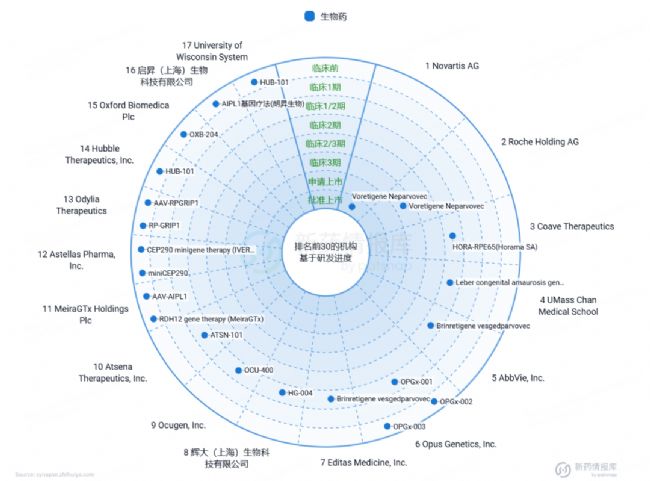
圖7 LCA基因治療藥物研發現狀
(圖片來源:智慧芽新藥情報庫)
RDDC助力LCA綜合征研究
罕見病數據中心(RDDC)是國內首個對外公開、免費的罕見病研究數據庫。
RDDC由罕見病基因治療聯盟理事長單位——清華珠三角研究院人工智能創新中心主持開發,并由副理事長單位賽業生物提供生物遺傳技術支持,歷經1.0至2.0版本升級,可為用戶提供相應罕見病的信息,并更好的服務科研人員對于數據查詢和數據挖掘的需求。
用戶登錄后,可以在短時間內完成從靶點基因發現,到靶點基因的表型和功能查詢,以及選擇獲得當前市場上和靶點基因表型最相關的動物模型,從而快速制定研究路線,開展針對疾病致病基因的科學研究和藥物發現工作,歡迎掃碼體驗!
*聲明:RDDC數據和工具僅為科研使用,僅供參考,不可作為醫學診斷和評判的最終定論。
參考文獻:
[1]T.C. Chen, D.S. Huang, C.W. Lin, C.H. Yang, C.M. Yang, V.Y. Wang, J.W. Lin, A.C. Luo, F.R. Hu, P.L. Chen, Genetic characteristics and epidemiology of inherited retinal degeneration in Taiwan, NPJ Genom Med 6(1) (2021) 16.
[2]I. Perrault, J.M. Rozet, S. Gerber, I. Ghazi, C. Leowski, D. Ducroq, E. Souied, J.L. Dufier, A. Munnich, J. Kaplan, Leber congenital amaurosis, Mol Genet Metab 68(2) (1999) 200-8.
[3]R.K. Koenekoop, I. Lopez, A.I. den Hollander, R. Allikmets, F.P. Cremers, Genetic testing for retinal dystrophies and dysfunctions: benefits, dilemmas and solutions, Clin Exp Ophthalmol 35(5) (2007) 473-85.
[4]E.M. Stone, Leber congenital amaurosis - a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture, Am J Ophthalmol 144(6) (2007) 791-811.
[5]R.K. Koenekoop, An overview of Leber congenital amaurosis: a model to understand human retinal development, Surv Ophthalmol 49(4) (2004) 379-98.
[6]N. Kumaran, A.T. Moore, R.G. Weleber, M. Michaelides, Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions, Br J Ophthalmol 101(9) (2017) 1147-1154.
[7]K.L. Heher, E.I. Traboulsi, I.H. Maumenee, The natural history of Leber's congenital amaurosis. Age-related findings in 35 patients, Ophthalmology 99(2) (1992) 241-5.
[8]S.G. Foxman, J.R. Heckenlively, J.B. Bateman, J.D. Wirtschafter, Classification of congenital and early onset retinitis pigmentosa, Arch Ophthalmol 103(10) (1985) 1502-6.
[9]M. Alkharashi, A.B. Fulton, Available Evidence on Leber Congenital Amaurosis and Gene Therapy, Semin Ophthalmol 32(1) (2017) 14-21.
[10]S. Hanein, I. Perrault, S. Gerber, G. Tanguy, F. Barbet, D. Ducroq, P. Calvas, H. Dollfus, C. Hamel, T. Lopponen, F. Munier, L. Santos, S. Shalev, D. Zafeiriou, J.L. Dufier, A. Munnich, J.M. Rozet, J. Kaplan, Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis, Hum Mutat 23(4) (2004) 306-17.
[11]S.R. Dharmaraj, E.R. Silva, A.L. Pina, Y.Y. Li, J.M. Yang, C.R. Carter, M.K. Loyer, H.K. El-Hilali, E.K. Traboulsi, O.K. Sundin, D.K. Zhu, R.K. Koenekoop, I.H. Maumenee, Mutational analysis and clinical correlation in Leber congenital amaurosis, Ophthalmic Genet 21(3) (2000) 135-50.
[12]Y. Chen, Q. Zhang, T. Shen, X. Xiao, S. Li, L. Guan, J. Zhang, Z. Zhu, Y. Yin, P. Wang, X. Guo, J. Wang, Q. Zhang, Comprehensive mutation analysis by whole-exome sequencing in 41 Chinese families with Leber congenital amaurosis, Invest Ophthalmol Vis Sci 54(6) (2013) 4351-7.
[13]A.I. den Hollander, R. Roepman, R.K. Koenekoop, F.P. Cremers, Leber congenital amaurosis: genes, proteins and disease mechanisms, Prog Retin Eye Res 27(4) (2008) 391-419.
[14]J.B. Ducloyer, G. Le Meur, T. Cronin, O. Adjali, M. Weber, [Gene therapy for retinitis pigmentosa], Med Sci (Paris) 36(6-7) (2020) 607-615.
[15]S.G. Jacobson, T.S. Aleman, A.V. Cideciyan, A. Sumaroka, S.B. Schwartz, E.A. Windsor, E.I. Traboulsi, E. Heon, S.J. Pittler, A.H. Milam, A.M. Maguire, K. Palczewski, E.M. Stone, J. Bennett, Identifying photoreceptors in blind eyes caused by RPE65 mutations: Prerequisite for human gene therapy success, Proc Natl Acad Sci U S A 102(17) (2005) 6177-82.



