
罕見遺傳性病法布雷病(FD)研究模型Gla-KO小鼠的介紹及應用
法布雷病(FD)是一種罕見的遺傳性疾病,屬于溶酶體貯存病(LSD)的一種。該病由溶酶體半乳糖苷酶A(α-GalA)活性缺乏引起,導致其代謝底物三己糖酰基鞘脂醇(GL3)和相關鞘糖脂在心臟、腎臟、胰腺、皮膚、肺和神經系統等人體各器官中大量貯積。這種貯積最終引發一系列相應的器官疾病,嚴重病例可能出現心腦血管并發癥或終末期腎病,甚至過早死亡[1]。然而,由于FD癥狀無特異性且罕見,通常在病情晚期才能被診斷出來,這給疾病的診斷、預防、治療和預后帶來了極大的挑戰。
法布雷病(FD)的遺傳學機制
法布雷病(FD)是由X染色體上的GLA基因發生突變引起的,這種突變導致溶酶體半乳糖苷酶A(α-GalA)的活性降低。α-GalA是三己糖酰基鞘脂醇(GL3)代謝過程中的關鍵酶,當α-GalA缺乏時,GL3會在全身細胞中逐漸積累,往往伴隨著早發中風、心律失常、心肌梗死或心力衰竭以及腎功能衰竭的高風險[1]。FD主要有兩種臨床表現形式:早發型(典型型)和衰減型(晚發型)。早發型FD的特征是α-GalA功能和活性幾乎完全喪失,導致早期出現廣泛的多器官并發癥,包括肢端感覺異常、出汗異常、角膜輪狀和血管角化瘤,以及心血管、腦血管和腎臟疾病,如心肌病、心律失常、中風和蛋白尿。而衰減型FD則由于α-GalA活性部分殘留,疾病在晚期出現,且表現各異,主要取決于剩余的α-GalA活性水平[1-2]。此外,由于致病基因位于X染色體上,FD在男性和女性患者之間存在顯著的性別差異,男性患者的癥狀通常比女性更嚴重。
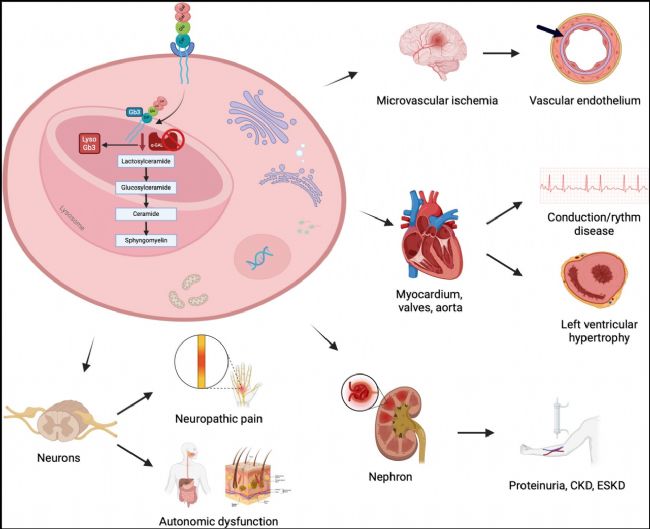
法布雷病(FD)病理生理學機制:不同細胞中鞘糖脂的溶酶體積累是法布雷病(FD)器官損傷的原因[2]
法布雷病(FD)療法開發
法布雷病(FD)的主要治療方法有兩種:一種是每周靜脈注射阿加糖酶α或阿加糖酶β的酶替代療法(ERT),另一種是每日口服米加司他(Migalastat)分子伴侶療法。然而,這些療法對疾病進展的影響因素眾多,且由于治療費用高昂(約25萬歐元/年),許多患者可能無法承受[3]。因此,深入研究FD的機制并開發新型有效的療法成為研究的重點。目前,包括Abeona Therapeutics、UniQure和Spark Therapeutics在內的10多家企業正在開發由腺相關病毒(AAV)載體介導的基因療法[4-5]。這類療法通過AAV載體將GLA基因的健康拷貝送入體內,然后基因進行轉錄翻譯,產生功能性α-Gal A酶。目前,至少有六項AAV-hGLA基因療法已進入臨床試驗階段,這為法布雷病(FD)的治療帶來了新的希望。
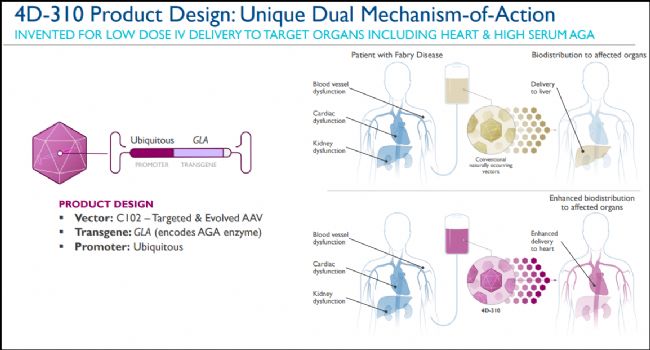
法布雷病(FD)AAV基因療法4D-310的治療策略[6]
法布雷病(FD)動物模型
AAV基因療法是法布雷病(FD)新療法的重要研究方向。在這些療法的開發過程中,首先需要對藥物的藥效和安全性進行動物體內評估。由于法布雷病(FD)主要由GLA基因突變導致α-Gal A酶的功能和活性缺失,Gla基因敲除小鼠(Gla-KO)因此成為這些療法臨床前評價的首選模型。目前,已有多個進入臨床階段的療法,包括Freeline的FLT190(2期)[7]、4DMT的4D-310(1/2期)[6, 8]、UniQure的AMT-191(1/2期)[9]和Sangamo的ST-920(1/2期)[10-12],都采用了Gla-KO小鼠進行藥效評估。此外,CANbridge和Genzyme等企業也在開發非臨床階段的AAV療法[13-14],并同樣采用Gla-KO小鼠進行評估。因此,Gla-KO小鼠被廣泛認為是法布雷病(FD)疾病模型的“金標準”,可用于研究疾病機制以及新療法的臨床前療效評估。
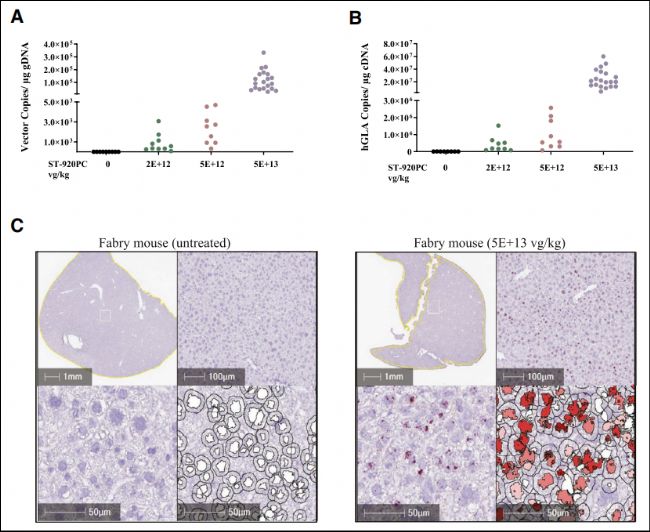
Gla-KO小鼠(Fabry mouse)用于AAV基因療法ST-920的臨床前評估[11]
賽業生物Gla-KO小鼠助力法布雷病(FD)研究
Gla-KO小鼠在多種細胞類型中會積累三己糖酰基鞘脂醇(GL3),這種現象隨著年齡的增長而加劇。其體內的組織學變化,包括底物的積累性質和時間影響,與法布雷病患者的病理生理過程有著極高的相似性[15]。因此,Gla-KO小鼠被廣泛應用于評估酶替代療法、AAV基因療法和底物減少療法的臨床前療效和安全性。賽業生物開發的Gla-KO小鼠(產品編號:S-KO-00955)同樣被廣泛用于機制研究和療法評估。例如,利用肝臟特異性AAV療法治療Gla-KO小鼠(Orphanet J Rare Dis, 2023)的研究[16],并利用其評估新型ERT療法效果以及藥物代謝(Biomolecules, 2022)[17]。此外,Gla-KO小鼠還助力溶酶體缺陷與先天性免疫之間的內在聯系的揭示和溶酶體貯存病(LSD)新治療策略的提出(Nature Cell Biology, 2024)[18]
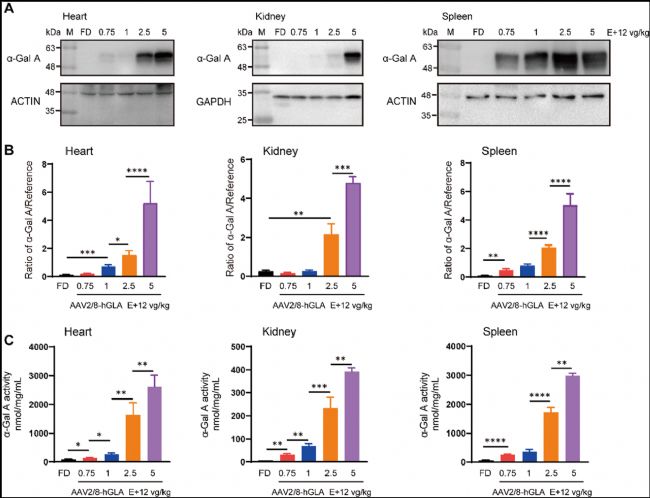
賽業生物Gla-KO小鼠(FD)助力AAV基因療法的臨床前研究[16]
[1] Chan B, Adam DN. A Review of Fabry Disease. Skin Therapy Lett. 2018 Mar;23(2):4-6.
[2] Lerario S, Monti L, Ambrosetti I, Luglio A, Pietra A, Aiello V, Montanari F, Bellasi A, Zaza G, Galante A, Salera D, Capelli I, La Manna G, Provenzano M. Fabry disease: a rare disorder calling for personalized medicine. Int Urol Nephrol. 2024 Apr 13.
[3] Lenders M, Brand E. Fabry disease - a multisystemic disease with gastrointestinal manifestations. Gut Microbes. 2022 Jan-Dec;14(1):2027852.
[4] Domm JM, Wootton SK, Medin JA, West ML. Gene therapy for Fabry disease: Progress, challenges, and outlooks on gene-editing. Mol Genet Metab. 2021 Sep-Oct;134(1-2):117-131.
[5] Rodríguez-Castejón J, Beraza-Millor M, Solinís MÁ, Rodríguez-Gascón A, Del Pozo-Rodríguez A. Targeting strategies with lipid vectors for nucleic acid supplementation therapy in Fabry disease: a systematic review. Drug Deliv Transl Res. 2024 Apr 8.
[6] 4D molecular therapeutics. An Open-label, Phase 1/2 Trial of Gene Therapy 4D-310 in Adult Males with Fabry Disease. Retrieved April 18, 2024, from 4DMT PPT Template (4dmoleculartherapeutics.com)
[7] Jeyakumar JM, Kia A, Tam LCS, McIntosh J, Spiewak J, Mills K, Heywood W, Chisari E, Castaldo N, Verhoef D, Hosseini P, Kalcheva P, Cocita C, Miranda CJ, Canavese M, Khinder J, Rosales C, Hughes D, Sheridan R, Corbau R, Nathwani A. Preclinical evaluation of FLT190, a liver-directed AAV gene therapy for Fabry disease. Gene Ther. 2023 Jun;30(6):487-502.
[8] Shen JS, Arning E, West ML, Day TS, Chen S, Meng XL, Forni S, McNeill N, Goker-Alpan O, Wang X, Ashcraft P, Moore DF, Cheng SH, Schiffmann R, Bottiglieri T. Tetrahydrobiopterin deficiency in the pathogenesis of Fabry disease. Hum Mol Genet. 2017 Mar 15;26(6):1182-1192.
[9] uniQure. Patient enrollment in the clinical trial of AMT-191, uniQure’s gene therapy candidate for the treatment of Fabry disease, is expected to begin in the first half of 2024. Retrieved April 18, 2024, from Fabry Disease | Programs & Pipeline | uniQure
[10] Pagant S, Huston MW, Moreira L, Gan L, St Martin S, Sproul S, Holmes MC, Meyer K, Wechsler T, Desnick RJ, Yasuda M. ZFN-mediated in vivo gene editing in hepatocytes leads to supraphysiologic α-Gal A activity and effective substrate reduction in Fabry mice. Mol Ther. 2021 Nov 3;29(11):3230-3242.
[11] Yasuda M, Huston MW, Pagant S, Gan L, St Martin S, Sproul S, Richards D, Ballaron S, Hettini K, Ledeboer A, Falese L, Cao L, Lu Y, Holmes MC, Meyer K, Desnick RJ, Wechsler T. AAV2/6 Gene Therapy in a Murine Model of Fabry Disease Results in Supraphysiological Enzyme Activity and Effective Substrate Reduction. Mol Ther Methods Clin Dev. 2020 Jul 9;18:607-619.
[12] Takahashi H, Hirai Y, Migita M, Seino Y, Fukuda Y, Sakuraba H, Kase R, Kobayashi T, Hashimoto Y, Shimada T. Long-term systemic therapy of Fabry disease in a knockout mouse by adeno-associated virus-mediated muscle-directed gene transfer. Proc Natl Acad Sci U S A. 2002 Oct 15;99(21):13777-82.
[13] PR Newswire. CANbridge Pharmaceuticals to Present Fabry Disease Gene Therapy Abstract at ESGCT 30th Annual Congress. Retrieved April 18, 2024, from CANbridge Pharmaceuticals to Present Fabry Disease Gene Therapy Abstract at ESGCT 30th Annual Congress (prnewswire.com)
[14] Ziegler RJ, Cherry M, Barbon CM, Li C, Bercury SD, Armentano D, Desnick RJ, Cheng SH. Correction of the Biochemical and Functional Deficits in Fabry Mice Following AAV8-mediated Hepatic Expression of α-galactosidase A. Mol Ther. 2007 Mar;15(3):492-500.
[15] Ohshima T, Murray GJ, Swaim WD, Longenecker G, Quirk JM, Cardarelli CO, Sugimoto Y, Pastan I, Gottesman MM, Brady RO, Kulkarni AB. alpha-Galactosidase A deficient mice: a model of Fabry disease. Proc Natl Acad Sci U S A. 1997 Mar 18;94(6):2540-4.
[16] Deng M, Zhou H, He S, Qiu H, Wang Y, Zhao AY, Mu Y, Li F, Zhao AZ. Systematic gene therapy derived from an investigative study of AAV2/8 vector gene therapy for Fabry disease. Orphanet J Rare Dis. 2023 Sep 5;18(1):275.
[17] Deng M, Zhou H, Liang Z, Li Z, Wang Y, Guo W, Zhao AY, Li F, Mu Y, Zhao AZ. Development of Lanzyme as the Potential Enzyme Replacement Therapy Drug for Fabry Disease. Biomolecules. 2022 Dec 27;13(1):53.
[18] Wang A, Chen C, Mei C, Liu S, Xiang C, Fang W, Zhang F, Xu Y, Chen S, Zhang Q, Bai X, Lin A, Neculai D, Xia B, Ye C, Zou J, Liang T, Feng XH, Li X, Shen C, Xu P. Innate immune sensing of lysosomal dysfunction drives multiple lysosomal storage disorders. Nat Cell Biol. 2024 Feb;26(2):219-234.



