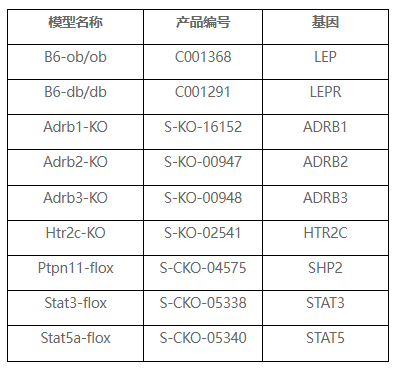
肥胖癥研究小鼠的介紹及應用之非LEP/LEPR遺傳模型
遺傳性肥胖主要由瘦素信號通路分子的異常導致,以該通路基因(主要是LEP和LEPR)為基礎構建的動物模型廣泛用于肥胖癥的研究。但除LEP/LEPR外,還有一些其它瘦素通路及調控通路基因同樣和肥胖的發生發展密切相關,可以用于肥胖癥模型構建和機制研究。

調節體重的瘦素信號通路及調控通路[1]
β-腎上腺素能受體(βAR)家族通路缺陷模型
在上世紀以ob/ob小鼠為主要的肥胖癥模型時,就有研究發現該小鼠β-腎上腺素能受體(βAR)信號存在缺陷,表現為脂肪分解缺乏和非顫抖性產熱受損,但已發現的β1AR和β2AR兩種亞型活性并無較大變化。后續的研究讓人們逐漸發現在脂肪組織中高表達的β3AR亞型,并對這三種亞型作了更精準的分類和定義[2]。在ob/o小鼠中,β3AR表達完全缺失,β1AR同樣大幅下降,提示βAR家族在抑制脂肪發育和肥胖發展的潛在作用[3]。
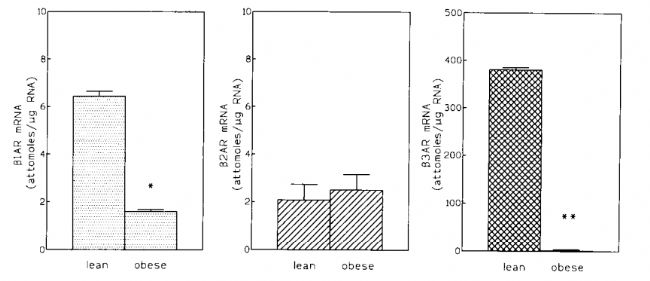
ob/ob小鼠體內β3AR表達完全缺失[3]
至此,研究揭示了三種βAR的不同作用,即負責調節心率和心肌收縮的β1AR,與平滑肌松弛、胰島素和胰高血糖素分泌和脂肪分解有關的β2AR,以及負責嚙齒動物中白色脂肪脂肪分解和棕色脂肪產熱作用的β3AR[4]。
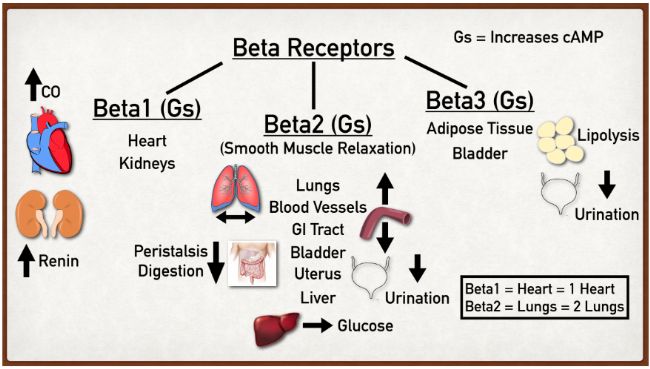
三種βAR的不同表達部位和作用[4]
β1AR、β2AR和β3AR都通過不同的途徑不同程度的抑制肥胖進展,作為脂肪中含量最多的亞型,β3AR促進脂肪組織分解、骨骼肌產熱、增加肌肉葡萄糖攝取量并減少肝臟葡萄糖輸出量,具有抗肥胖和抗糖尿病作用。相反,敲除β3AR(Adrb3基因編碼)的小鼠體內脂肪積累和食物攝入量均增加,高脂喂養會加劇肥胖表型,表現為體脂總量增加、蛋白質含量減少和無脂干重減少等,表明β3AR的缺失導致肥胖癥發生[5-6]。
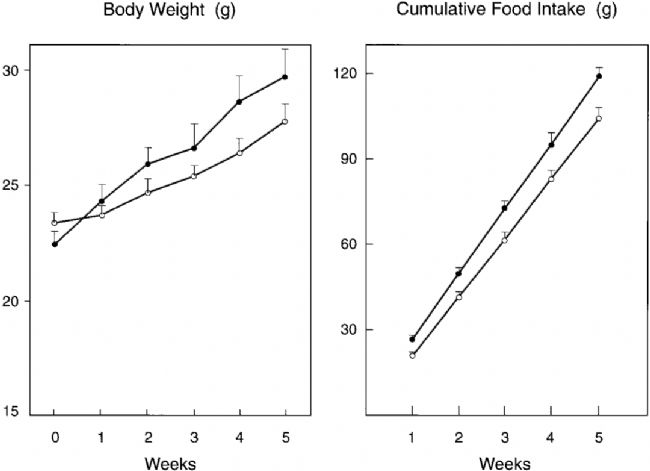
與野生型小鼠(○)相比,β3AR缺失小鼠(●)表現為體重和食物攝入量增加[5]
β1AR和β2AR主要負責心率和心肌收縮以及平滑肌松弛、胰島素和胰高血糖素分泌和脂肪分解的調控,對于肥胖進展的調控不是很明顯。但β1AR和β2AR同樣是飲食誘導產熱的必要條件,在機體抵御飲食誘導肥胖的過程中發揮著關鍵作用。同時缺乏三種亞型的小鼠表現為更嚴重的肥胖,包括棕色脂肪組織形態異常、耗氧量減少、適應性產熱受損、體重增加、棕色脂肪細胞大小增加和脂肪總量增加[7]。
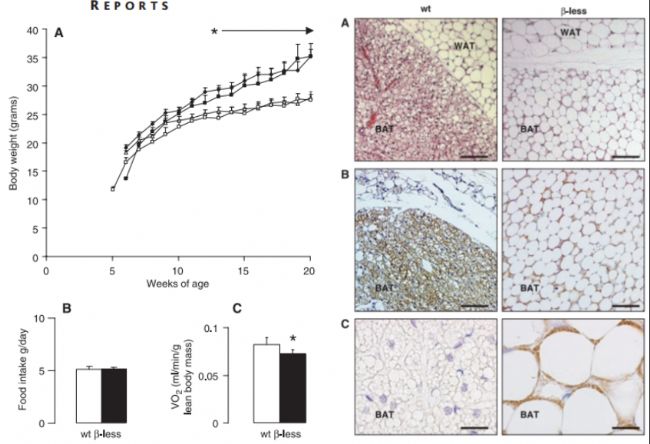
β1AR/β3AR/β2AR敲除小鼠(β-less)存在嚴重肥胖表型[7]
5-羥色胺/5-羥色胺2C受體通路缺陷模型
5-羥色胺(5-HT)在人體內發揮著復雜且多方面的作用,其主要功能是穩定情緒,同時也在消化系統和睡眠周期中發揮作用。5-HT2C受體是5-HT受體亞型之一,與5-HT共同在食物攝入和體重控制方面發揮作用[8]。貝勒醫學院等機構的研究顯示,5-HT2C受體基因(HTR2C)的突變在肥胖和適應不良行為中起著重要作用。一些嚴重肥胖患者攜帶罕見的HTR2C基因功能缺失型(LOF)突變,轉入人類HTR2C基因LOF突變的小鼠也發展為食欲過盛和肥胖[9]。

攜帶HTR2C功能缺失突變的小鼠出現貪食性肥胖[9]
此外,敲除小鼠體內Htr2c基因的表達,同樣可以導致進食過多、多動和肥胖,以及對厭食性5-HT藥物反應減弱等表型,在POMC神經元中重新表達Htr2c基因可減輕這些癥狀[10]。
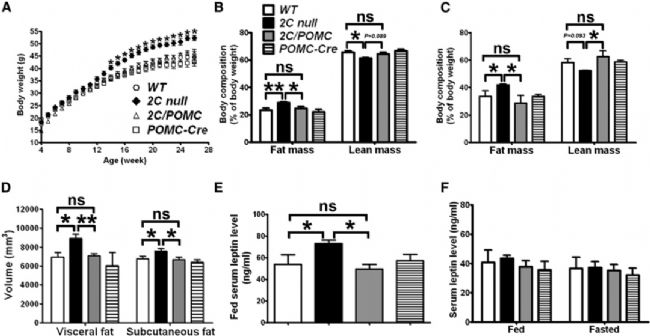
HTR2C-KO小鼠(2C null)體重和脂肪比例增加[10]
SHP2/ERK信號缺陷模型
瘦素對能量平衡的效應是通過激活瘦素受體的長形式(LR)實現的,刺激LR會導致STAT3磷酸化、Src同源-2酪氨酸磷酸酶(SHP2)和胰島素受體底物2(IRS2)激活。SHP2由Ptpn11基因編碼,通過調節RAS/ERK等信號通路來調節細胞的生長、分化和凋亡。中樞神經的SHP2信號介導瘦素的抗肥胖作用,Zhang等人的研究表明,前腦神經元條件性Shp2敲除小鼠(CaMKIIα-Cre;Shp2flox/flox)表現為肥胖,并具有代謝綜合征的多個特征,表明SHP2信號在調節能量平衡和代謝中發揮的重要作用[11]。而在POMC神經元中敲除Ptpn11同樣會導致小鼠(Ptpn1loxP/loxP;POMC-Cre)出現體重和脂肪含量的增加[12]。
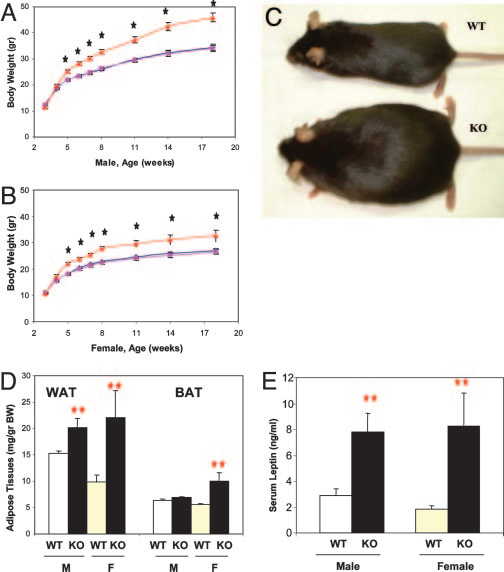
前腦神經元中Shp2條件性的敲除將導致小鼠嚴重肥胖[11]
此外,通過將泛神經元Cre小鼠(CRE3)與Shp2flox/flox小鼠雜交,產生大腦神經元特異性Shp2缺失的小鼠出現更嚴重的肥胖和糖尿病,并伴有高血糖、高胰島素血癥、高瘦素血癥、胰島素和瘦素抵抗、血管炎和糖尿病腎病等多種并發癥[13],為闡明人類肥胖和糖尿病以及并發癥發生的分子機制提供重要參考。
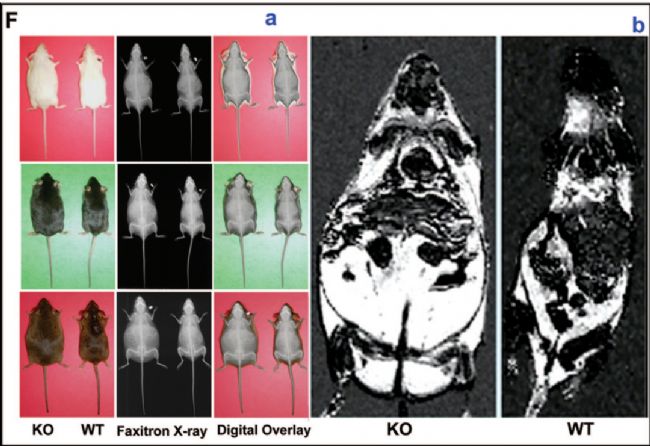
泛神經元條件性Shp2敲除小鼠呈現嚴重肥胖表型[13]
JAK2-STAT3/STAT5信號缺陷模型
瘦素與LepRb結合激活JAK2,從而導致LepRb在Tyr1138和Tyr1077位點的磷酸化。磷酸化的Tyr1138和Tyr1077與Src同源2(SH2)結構域結合,激活STAT3和STAT5。激活后的STAT3/STAT5轉到細胞核并作為轉錄因子調節靶基因的表達而起到調節的作用。神經元中STAT3 Tyr1138位點破壞會導致小鼠攝食過多和肥胖,與db/db小鼠的表型相似[14]。同樣的,STAT5 Tyr1077位點的破壞或大腦條件性Stat5敲除的小鼠都會出現多食和肥胖的表型[15]。
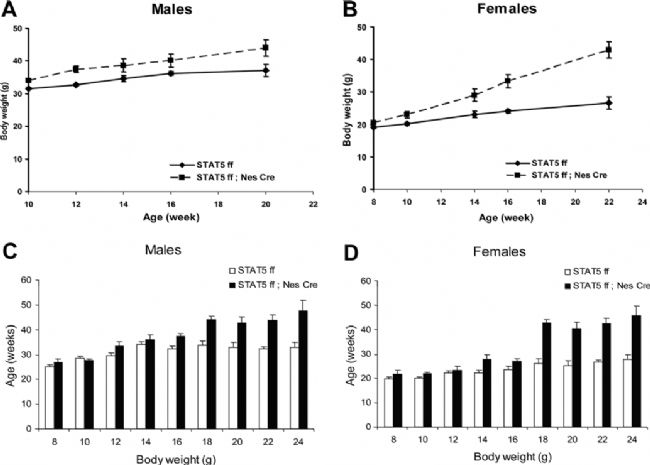
大腦特異性Stat5敲除導致小鼠體重上升[15]
肥胖癥研究模型推薦
小鼠基因編輯模型在肥胖和其它代謝疾病機制研究和藥物研發評價中起著重要作用,賽業生物擁有數千種自主研發的基因編輯小鼠品系,可提供包括LEP、LEPR、ADRB3和SHP2等在內的多種基因敲除或條件性敲除小鼠模型。同時也可根據您的科研需求進行專業化的定制服務,加速您的課題研究。