
漫長的“漸凍癥”肯尼迪病(KD)的發病機制與臨床表現介紹
當你正值壯年,遭遇全身“漸凍”,爬樓變得吃力,速度越來越慢,手指常常不聽使喚,夾菜都沒勁的時候一定要注意了,很有可能是肯尼迪病。肯尼迪病和漸凍癥(肌萎縮側索硬化)一樣都屬于運動神經元病。漸凍癥患者往往在3-5年內就無法活動、呼吸,而肯尼迪病的病情發展會緩慢一些,可以理解為是一種漫長的漸凍癥,雖然并不致命,但依然面臨手腳漸漸無力的事實。

什么是肯尼迪病[1]
肯尼迪病(Kennedy’s disease, KD),又稱為脊髓延髓肌萎縮癥(Spinal and bulbar muscular atrophy, SBMA),是一種成人發病的X連鎖隱性遺傳的神經系統變性疾病。最初由Kennedy等于1968年報道。
KD的病因是位于Xq11-12的雄激素受體 (androgen receptor, AR)基因第一外顯子一段CAG重復序列延長,導致其編碼的AR中一段多聚谷氨酰胺鏈(poly glut amine tract, Poly Q)延長。KD與HD(亨廷頓病)、齒狀核紅核黒質萎縮、SCA(遺傳性小腦共濟失調)1,2,3,6,7,17型同屬于Poly Q病。與其他Poly Q病不同,KD是唯一的X連鎖隱性遺傳疾病。KD的發病率約為2.5/100000。它屬于晚發疾病,患者多為中年男性,臨床表現為緩慢進展的對稱性肢體近端和球部的肌肉萎縮、無力,伴有“男性乳腺發育”等雄激素功能減退癥狀。KD患者發病后的平均生存期約為22~27年。在疾病發展過程中,患者生活質量會受到嚴重影響[2]。
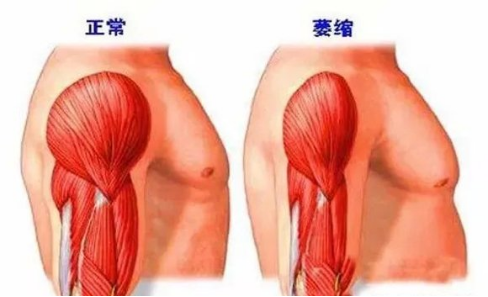
肯尼迪病肌肉狀態[3]
發病機制
肯尼迪病發病的分子基礎是AR的第一外顯子三核苷酸CAG異常擴增,導致其基因編碼的多聚谷氨酰胺異常聚集。正常人AR的CAG重復范圍從11到35,而肯尼迪病患者為40至62。
肯尼迪病是CAG多聚谷氨酰胺病的家族成員之一,通過對其他多聚谷氨酰胺有關神經變性疾病[包括亨廷頓病和幾種形式的脊髓小腦性共濟失調(SCA)]的觀察,CAG重復次數與疾病嚴重程度和發病年齡呈負相關,即異常擴增的倍數越多,發病時間越早。在肯尼迪病的神經傳導研究中,CAG重復次數和發病年齡在運動為主和感覺為主的患者顯著不同,表明CAG重復次數較多與運動為主的類型相關,而CAG重復次數較少與感覺為主的類型明顯相關[4]。
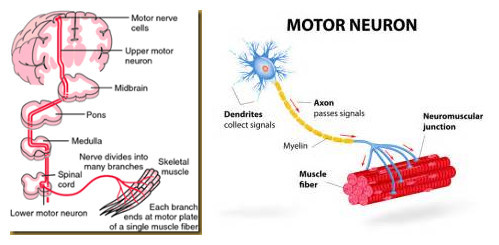
肯尼迪病的病理機制[1]
臨床表現
1、常規化驗
肯尼迪病患者血清肌酸肌酶和乳酸脫氫酶出現不同程度的升高,性激素如睪酮,黃體酮,促卵泡激素,黃體生成素水平也可能出現異常,不過腰穿腦脊液檢查通常正常。某些患者可出現高脂血癥以及糖耐量受損。
2、電生理檢查:
神經傳導檢查可提示感覺神經動作電位波幅降低,感覺神經傳導速度減慢。針極肌電圖多呈廣泛神經源性損害,存在進行性和(或)慢性失神經改變,出現多個自發電位,運動單位動作電位時限顯著增寬,甚至出現巨大電位,大力收縮時呈單純相。單纖維機電圖(SFEMG)上顫抖(jitter)值明易增寬,運動單位計數也明顯減少。
3、肌肉活檢:
主要表現為神經源性損害,有時可合并肌源性損害特征。
4、神經活檢:
腓腸神經活檢可見大的有髓纖維減少,少量纖維脫髓鞘,施萬細胞變性。
5、基因檢測:
既往多數文獻把CAG拷四次數大于40次作為確診標準。2011年歐洲神經科學聯合會指南將CAG重復序列數日≥35次作為診SBMA的依據。CAG異常擴增長度與發病年齡和起病癥狀有關,與癥病的進展無關。與其他突變基因重復擴增的癥病相似,SBMA亦呈現“遺傳早現”現象,重復拷貝數在傳代過程中不斷增加,導致發病時間逐代提前,癥狀逐代加重[7]。

一位肯尼迪病患者基因檢測AR基因CAG重復次數為41次[6]
參考文獻及引用圖片來源:
[1]https://zhuanlan.zhihu.com/p/30578113
[2]魯明. 肯尼迪病的研究進展[C]//.中華醫學峰會暨中華醫學會神經病學分會第八屆全國中青年神經病學學術會議論文匯編.[出版者不詳],2015:47-48.
[3]https://tieba.baidu.com/p/7279946603
[4]馬俊芳,崔麗英,崔博.肯尼迪病的臨床特點、發病機制和治療進展[J].中華神經科雜志,2015,48(04):344-347.
[5]Grunseich C, Rinaldi C, Fischbeck KH. Spinal and bulbar muscular atrophy: pathogenesis and clinical management [J ].Oral Dis, 2014, 20 (1) : 6-9.
[6]俞立強,方琪,蔣覺安,許麗珍.肯尼迪病臨床、病理及遺傳學特點[J].臨床神經病學雜志,2015,28(04):296-298.
[7]https://www.nrdrs.org.cn/app/rare/disease-list-article.html?index=109



