
遺傳性脊髓小腦共濟失調的介紹及治療方式
在對一個進行性共濟失調患者進行遺傳性脊髓小腦共濟失調(spino cerebellar ataxia, SCA)診斷之前,應通過詳細的病史詢問、體格檢查、實驗室檢查等除外獲得性的病因,因SCA的臨床癥狀有很大的重疊性及異質性,故確診主要依靠基因檢測。SCA的診斷依據如下:
①以進行性共濟失調為主要臨床表現,并除外獲得性病因導致的共濟失調;
②上一代有類似疾病的病史;
③SCA基因檢測呈陽性[1]。
如果診斷SCA的臨床證據明確,則應啟動分子遺傳學檢測。只有在特殊情況下,如已知家族中特定的SCA基因型、臨床表型高度提示某一種SCA(例如SCA7中的視力喪失)或某一種SCA的地區流行率較高(例如古巴的SCA2),才建議進行有針對性的單一基因檢測。而在其他情況下,多推薦采用系統的SCA基因檢測。首先要進行多聚谷酰胺SCA的動態突變檢測(即SCA1、SCA2、SCA3、SCA6、SCA7、SCA17、DRPLA),因為此類SCA患病率相對較高。當多聚谷酰胺的CAG動態突變檢測為陰性時,可根據現實情況選擇適宜順序:
①全外顯子測序,可檢測傳統突變類型的SCA,也可以在發現新的共濟失調基因時進行重新分析;
②WES不能檢測到重復突變,為了評估SCA的所有遺傳原因,則必須對非翻譯區的重復突變(即SCA8、SCA10、SCA12、SCA31、SCA36和SCA37)進行動態突變的檢測;
③如果全外顯子測序未包含對于具有特定缺失的搜索,則需對SCA15/SCA16的ITPR1中大的缺失進行特定檢測。由于基因檢測的成本在不斷下降,這里討論的復雜基因測試策略很可能會被全基因組測序所取代,但目前全基因組測序還不能作為常規檢測手段。
沒有SCA家族史的共濟失調患者,也可能由于自發突變、突變外顯性降低或錯誤的親子關系而有遺傳性病因,如通過仔細的病史詢問、實驗室檢查,包括MRI和神經速度傳導等,已除外獲得性病因的患者則應該進行合理的基因檢測。對美國和歐洲的散發性成人型共濟失調患基因篩查研究發現,15%-24%的患者SCA相關基因發生突變。SCA6是最易漏診的多聚谷氨酰胺SCA,因為SCA6是遲發性SCA最常見的類型,傳遞SCA6基因的親代可能在患者發病前就已經死亡。對于散發性共濟失調患者,以上基因檢測策略同樣適用,而Friedreich共濟失調作為最常見的常染色體隱性遺傳共濟失調,也可表現為散發性共濟失調,雖然同樣是由三核苷酸重復造成的,但很容易通過動態基因檢測鑒別。
動物模型
2015年有團隊構建了人SCA3敲入小鼠模型,用人突變ATXN3cDNA的8-11外顯子編碼區及3’UTR區域替換鼠Atxn3基因組DNA的8-11號外顯子及3’UTR區域,人突變ATXN3帶有91個CAG重復序列,該小鼠模型為SCA3-Ki91模型在SCA3-Ki91模型中,他們發現突變ATXN3蛋白在12月月齡的SCA3-Ki91小鼠模型的全腦組織均有表達,并且形成核內包涵體,小腦浦肯野細胞退行性改變伴有星形膠質細胞增生。同時CAG重復序列的傳代不穩定性在SCA3-Ki91小鼠模型中也有體現。
另一個團隊在小鼠10號外顯子CAG重復序列區域插入82個CAG重復片段,構建了雜合Atxn3Q82小鼠,小鼠模型的病理發現和SCA3-Ki91類似,在小鼠的神經元內發現大量的突變ATXN3蛋白的核內包涵體,并且呈組織特異性。
后來又有團隊用細菌人工染色體(Bacteria artificial chromosome, BAC)構建SCA3BAC轉基因小鼠模型,帶有全長的人突變ATXN3基因,包括全部外顯子、內含子,以及上游的啟動子和其他調控元件。在BAC-SCA3首建鼠到達7個月月齡時,它的全腦組織進行重量檢測和免疫熒光檢測。BAC-SCA3雄性首建鼠A的體重和腦組織重量都比同籠同月齡的雌鼠小,說明該首建鼠出現了嚴重的體重下降和腦萎縮。紅色方框里面顯示的是小鼠的小腦組織;在免疫熒光檢測結果可以看出BAC-SCA3小鼠在7個月時,與野生型小鼠相比,小腦、腦干和海馬皮層有顯著的ATXN3蛋白聚集現象,以小腦更為顯著,然而在大腦皮層和基底節并未檢測到明顯的ATXN3。SCA3患者受累腦區也以小腦和腦干為主,皮層和基底節受累較少。該結果說明BAC-SCA3小鼠的受累腦區和SCA3患者的受累腦區較為一致[2]。

表1 月齡BAC-SCA3首建小鼠與野生型小鼠體重及腦組織重量的比較[2]
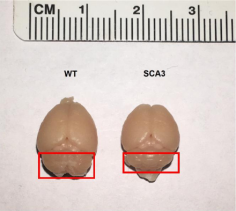
7月齡BAC-SCA3首建小鼠A與野生型小鼠的腦體積大小的比較[2]
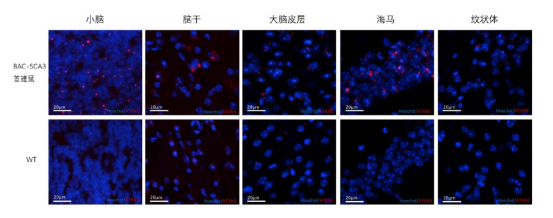
BAC-SCA3首建鼠A和野生型小鼠不同腦區ATXN3蛋白的聚集。藍色熒光為細胞核染色,紅色熒光為ATXN3蛋白。上為BAC-SCA首建鼠A,下組為野生型小鼠[2]
治療方式
目前SCA缺乏有效的治療方法,臨床上主要以對癥支持治療為主。隨著近年來對SCA病理生理機制研究的不斷深入,臨床發現了反義寡核苷酸(ASO)、小干擾RNA(siRNA)等新的治療靶點,但均未進入臨床試驗[1]。
藥物治療
目前還沒有藥物被批準用于SCA的常規治療。對于共濟失調癥狀,一些小型研究已經對多種藥物進行了研究。
①抗氧化劑:輔酶Q10和維生素E、艾地苯醌等。
②神經營養藥物:三磷腺苷、肌酐、維生素B族等。
③交感神經藥物抑制劑:安非他明、莫達非尼等。
④改善代謝治療:吡拉西坦、左旋肉堿、鐵螯合劑、鹽酸多奈哌齊等。
⑤其他藥物:人促紅細胞生成素、鉀通道阻滯劑、環絲氨酸等[3]。
支持治療
①物理治療:SCA患者發病早期即可進行物理治療,以便早期制定維持正常功能(肢體平衡、協調、正常姿勢)的策略和防止跌倒。
②職業治療:當患者日常活動越來越困難時,推薦職業治療師的介入,干預應側重于滿足患者及照顧者的功能目標和職業需求。
③語言及言語治療:語言治療師根據病情通過聲學儀器、指引及重復性練習,或利用視覺及聽覺輔助等方法給予治療。
基因治療
基因治療是通過各種方式減少變異擴增的polyQ蛋白表達水平,主要方法包括運用反義寡核苷酸(anti sense oligonucleotide, ASOs),DNA編輯技術CRISPR/CRISPR-Cas9或是通過RNA干擾敲除改變polyQ基因。同樣ASOs經腦室注射入不同的SCA2模型小鼠,發現SCA2小鼠的運動癥狀有改善并且降低了小腦中ATXN2表達,小腦ataxin-2蛋白減低。除了運用ASOs,還可以通過RNA干擾抑制致病基因的表達,有研究用RNA干擾抑制在SCA3小鼠模型中的ATXN3,顯示可以提高其運動功能。CRISPR/Cas9可能在polyQ SCAs的臨床試驗中具有潛力,但是包括基因組不穩定等問題仍需要解決。現階段運用各種基因治療polyQ SCAs的方法都在臨床前試驗階段,隨著研究的不斷深入,基因治療有潛力從病因方面治療polyQ SCAs,并對各亞型SCAs進行疾病修飾治療[4]。
干細胞移植治療
基因治療尚在研究階段,原理多為選擇性抑制SCAs致病基因表達。干細胞移植手術已在國內臨床上相繼開展,其基本原理為誘導多能干細胞分化為神經干細胞,替換受損細胞,并通過旁分泌作用為神經傳導提供更好的微環境。但其有效性和安全性還有待于進一步觀察研究[3]。
神經調節治療
神經調節治療主要包括深部腦刺激(deep brain stimulation, DBS),經顱磁刺激 (transcranial magnetic stimulation, TMS)以及經顱直流電刺激(transcranial direct current stimulation, tDCS),除了DBS需要經手術有創安裝電極,其余均是無創的。近年來,多項研究顯示神經調節可能對改善共濟失調的癥狀有幫助[4]。
RDDC助力罕見病研究
罕見病數據庫(以下簡稱“RDDC”)由罕見病基因治療聯盟理事長單位——清華珠三角研究院人工智能創新中心主持開發,并由副理事長單位賽業生物提供生物遺傳技術支持,歷經1.0至2.0版本升級,可為用戶提供相應罕見病的信息,并更好的服務科研人員對于數據查詢和數據挖掘的需求。
用戶登錄后,可以在短時間內完成從靶點基因發現,到靶點基因的表型和功能查詢,以及選擇獲得當前市場上和靶點基因表型最相關的動物模型,從而快速制定研究路線,開展針對疾病致病基因的科學研究和藥物發現工作,歡迎掃碼體驗!
*聲明:RDDC數據和工具僅為科研使用,僅供參考,不可作為醫學診斷和評判的最終定論。
賽業生物基因治療一站式解決方案
近年來全球多款基因治療藥物相繼獲批上市,還有多種針對不同適應癥的基因治療藥物正處于臨床研究階段,基因治療已然為眾多亟待拯救的患者帶來新的治療希望!
賽業生物基因治療一站式解決方案可為從事基因治療的研究者提供更高效的基因功能解析與基因治療一站式整體解決方案,包括靶點篩選與功能研究,動物模型構建和病毒載體如AAV、LV、ADV等設計與包裝,以及表型分析等全流程服務。
參考文獻及引用圖片來源:
[1]吳方瑞,鐘敏.脊髓小腦性共濟失調的臨床表現、發病機制及診療研究進展[J].山東醫藥,2021,61(23):112-115.
[2]杜依楚. 中國人群脊髓小腦性共濟失調3型的臨床研究及新型BAC-SCA3轉基因小鼠模型的構建[D].浙江大學,2020.DOI:10.27461/d.cnki.gzjdx.2020.002464.
[3]王東浩,黃艷梅,楊保勝.脊髓小腦性共濟失調的基因診斷及治療研究進展[J].新鄉醫學院學報,2016,33(07):639-641.
[4]孫迪,胡興越.多聚谷氨酰胺脊髓小腦性共濟失調的治療研究進展[J].全科醫學臨床與教育,2021,19(04):351-354.DOI:10.13558/j.cnki.issn1672-3686.2021.004.020.



