
特異性靶向治療杜氏肌營養不良癥(DMD)的新型療法的介紹
近日,在美國FDA召開的專家咨詢會議中,針對Sarepta Therapeutics和羅氏(Roche)聯合開發的SRP-9001這一基因療法,FDA細胞、組織和基因治療咨詢委員會(CTGTAC)以8:6的微弱投票優勢對其通過加速審批通道上市予以支持。作為一款特異性靶向治療杜氏肌營養不良癥(DMD)的新型療法,SRP-9001通過AAV遞送編碼基因,促使微肌營養不良蛋白增加以改善患者的肌肉功能。

盡管SRP-9001已獲得委員會的支持,但最終審查結果猶未可知。FDA可能存在的顧慮在哪里?其安全性和有效性到底能否得到認可?
這里我們從臨床前研究的角度出發,圍繞以下幾個要點,一起走近DMD基因治療及其相關疾病模型:
1.關于杜氏肌營養不良癥
2.代表性基因療法及相關模型:AAV補充mini基因、ASO跳躍外顯子、CRISPR基因編輯
3.針對疾病模型短板可采用怎樣的創新策略
關于杜氏肌營養不良癥
杜氏肌營養不良癥是由DMD突變導致的抗肌萎縮蛋白功能異常,最終造成肌肉進行性壞死的X染色體隱性遺傳性罕見病。其發病率約為36/10萬,中國約有6-10萬患者,屬于罕見病中的常見病,是全球最常見的肌營養不良類型。
DMD代表性基因療法及相關模型
DMD的基因治療主要分為三個類型:通過AAV補充mini dystrophin或micro-dystrophin;ASO跳躍外顯子療法;CRISPR基因編輯。
通過AAV補充mini dystrophin或micro-dystrophin
如果能起到作用的話,這種療法所適用的患者范圍有可能是最廣的,因為補充療法對不同突變情況的患者都可能起作用。Sarepta、Roche、Solid等公司正在布局這類AAV遞送mini dystrophin或micro-dystrophin療法,其中屬SRP-9001進展最快。
該管線的臨床前動物實驗用到的是DMD疾病研究經典模型mdx[1],mdx小鼠23號外顯子的CAA密碼子突變為TAA終止密碼子進而導致基因表達異常。mdx小鼠雖然出現了DMD的疾病表型,但相對于患者的實際情況來說:
(1)mdx小鼠表型更輕微,特別是在纖維肌溶解、肌肉萎縮方面;
(2)mdx小鼠的壽命大概是正常小鼠的80%,而DMD患者的壽命平均只有健康人群的1/3;
(3)mdx小鼠突變位點在E23,并非熱點突變位置,不太符合疾病同源性。
另外,mdx小鼠的不同背景表型還存在有些許不同的情況,這使得在mdx小鼠上進行研究需要考慮更多因素。
ASO跳躍外顯子療法
作為DMD相關基因治療管線中數量最多的一種,ASO跳躍外顯子療法得益于ASO的靶向性強、易于合成等優勢,Sarepta、Nippon、DYNE等藥企都有所布局,靶向跳躍的外顯子有44、45、50、51、53等,且文獻中也出現多個外顯子跳躍療法。
Nippon的管線NS-065/NCNP-01通過ASO靶向跳躍外顯子53,臨床前動物實驗使用了mdx52小鼠[2],該小鼠Dmd基因的52號外顯子缺失,導致在E53上終止翻譯。mdx52小鼠相對于mdx小鼠來說,雖然突變位點更接近于患者的熱點突變位置,更適合ASO基因治療,但在鼠源Dmd基因上篩選用于靶向人DMD基因的特異性ASO是存在一定風險的,因為人和小鼠之間存在一定的DMD基因序列差異。
而mdx小鼠非常不適合用于ASO跳躍療法,特別是ASO藥物特異性跳躍23號之后的外顯子的情況下,因為mdx小鼠在E23上已經有終止子,而跳躍其后的外顯子無法帶來行為學方面的藥效反應,僅通過mRNA表達水平來評估藥效是匱乏的。
有的文獻還使用了hDMDΔ52/mdx模型檢測ASO跳躍療法的藥效[3],該模型是通過TG轉入人全長DMD并敲除E52導致閱讀框破壞,然后與mdx小鼠進行雜交而構建的。hDMDΔ52/mdx模型雖然具有人DMD全長蛋白,但其DMD基因插入位置未知,不是X染色體,且需要進一步與mdx小鼠雜交,模型構建比較復雜。
CRISPR基因編輯療法
該療法在DMD疾病上的進展比較緩慢,其中一個原因是由于缺乏可以滿足特異性藥物靶點篩選及藥物預見性的DMD疾病模型。CRD-TMH-001是第一個基于CRISPR治療DMD的管線,目前已獲批IND。
在文獻中也出現不少CRISPR治療DMD的成功案例,如通過CBE胞嘧啶編輯器去除終止子,使得Dmd基因可以繼續翻譯,該實驗使用了Δ50;h51KI模型[4]。該模型是將小鼠Dmd的E51進行人源化,敲除E50并在E51上引入終止子。相對于mdx52,CRISPR編輯療法使用的模型Δ50;h51KI對藥物靶向序列的人源化要求程度更高,這也是CRISPR基因編輯療法所必須要考慮的重要因素,因為脫靶給患者帶來的負面影響是不可估量的。但同時,與hDMDΔ52/mdx相比,Δ50;h51KI模型具有人源化區域不足的缺點。
強勢破局的人源化小鼠
鑒于DMD基因的突變特點及現有模型的缺點,如構建復雜、Tg導致基因插入位點不確定、人源化區域不足和突變非熱點突變等問題,賽業生物自主研發了DMD疾病模型,除了經典模型mdx小鼠,還有DMD(hE8-30)、DMD(hE44-45)、DMD(hE49-53)等DMD熱點突變區域人源化小鼠。另外,我們可以通過在野生型人源化小鼠的基礎上進行基因再修飾,進一步構建熱點突變人源化疾病模型,為研究提供更理想的對照組和實驗組。
賽業生物hDMD小鼠特點:
● 擁有熱點突變區域的野生型及在此基礎上構建的點突變人源化疾病模型;
● 可以直接在已有的野生型人源化基礎上定制不同的點突變,提高效率及成功率;
● 人源化區域包括大部分藥物靶向區域,更適合可用于藥物篩選及藥效研究,特別是ASO、CRISPR、siRNA等基因治療相關藥物;
●人DMD基因原位插入,拷貝數確定,可穩定遺傳。

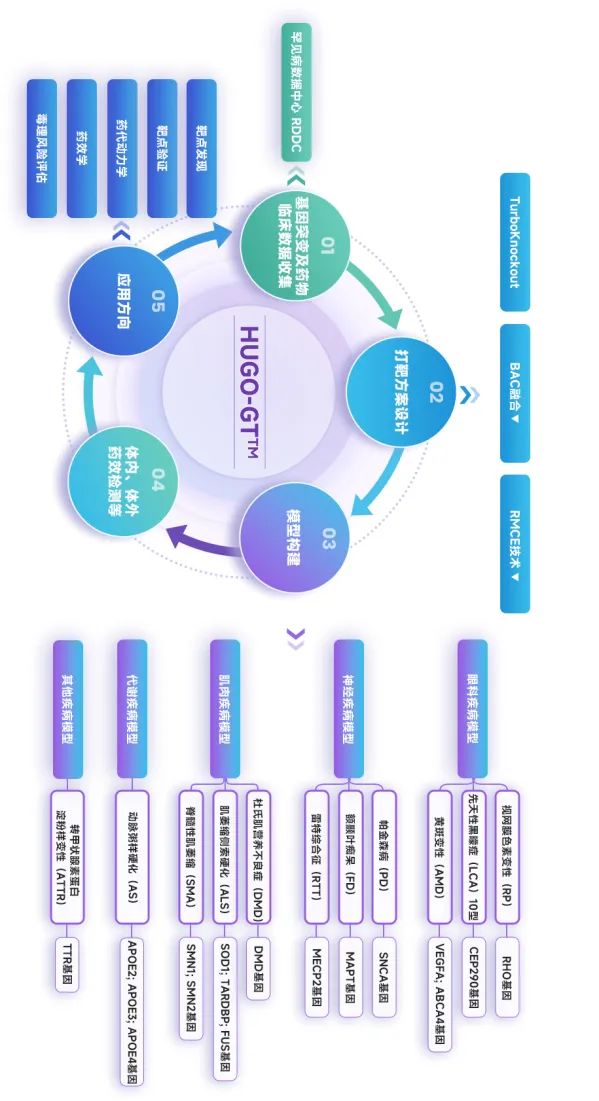
HUGO-GTTM全基因組人源化模型構建計劃
不僅是DMD,針對視網膜色素變性(RP)、黃斑變性(AMD)、帕金森病(PD)等多種疾病類型,如果要深入研究其致病機理,長片段甚至全基因組人源化小鼠是更好的選擇。但全基因組替換所需的技術難度高,大規模引入的外源序列可能會影響原本基因的表達調控。
為此,賽業生物啟動了HUGO-GTTM計劃,基于自主研發的TurboKnockout-Pro技術,對鼠源基因實現原位替換,成功構建了涵蓋更豐富干預靶點的全基因組人源化小鼠。HUGO-GTTM小鼠搭載了更高效的大片段載體融合技術,可以作為萬能模板進行針對性的突變定制服務,是更貼近真實世界生物機制的藥物臨床前研究模型。
[1] Ryder-Cook AS, Sicinski P, Thomas K, et al. Localization of the mdx mutation within the mouse dystrophin gene. [J]. EMBO,1988.
[2] Mizobe Y , Miyatake S , Takizawa H , et al. In Vivo Evaluation of Single-Exon and Multiexon Skipping in mdx52 Mice[J].Methods Mol Biol, 2018.
[3] Hoen P A C ' , Meijer E J D , Boer J M , et al. Generation and Characterization of Transgenic Mice with the Full-length Human DMD Gene[J]. Journal of Biological Chemistry, 2008, 283.
[4]Zhang Y, Li H, Nishiyama T, McAnally JR , et al. A humanized knockin mouse model of Duchenne muscular dystrophy and its correction by CRISPR-Cas9 therapeutic gene editing[J]. Mol Ther Nucleic Acids. 2022.



