
KDM2b基因與KDM2b基因敲除小鼠模型介紹

賽業生物《每周一鼠》,每周五更新,為大家講解一個小鼠模型的故事,希望對大家了解不同的小鼠模型有所幫助。今天和大家見面的是KDM2b基因敲除小鼠。
KDM2b基因
KDM2B(又名JHDM1B、FBXL10、NDY1)定位于細胞核內,是組蛋白H3K4me3和H3K36me2的特異性去甲基化酶。KDM2B可以調控細胞凋亡,抑制細胞的衰老,調控造血干細胞的自我更新、神經管的形成以及胚胎的發育,此外它還參與了腫瘤的發生發展。KDM2B屬于含有亮氨酸的重復結構Fbls類。已發現Kdm2b存在多個選擇性剪接轉錄,因此一些轉錄本的功能仍沒被完全確定[1]。
與KDM2b相關的疾病包括Cohen綜合征和嬰兒痙攣癥。該蛋白相關作用途徑包括染色質三維結構的調控和染色質表觀遺傳修飾。與該基因相關GO注釋的功能包括rRNA結合能力和組蛋白去甲基化酶活性。該基因的一個重要同源基因是KDM2A[2]。
KDM2b敲除小鼠模型
1●該基因不同轉錄本缺失小鼠
產生不同表型:
一項研究發現,該基因(長轉錄本)缺失的等位基因純合小鼠出生后大腦裸露、胚胎期或出生后死亡、眼組織異常、卷尾、少精子癥、細胞凋亡增加和神經元前體增殖增加[3]。
之前的研究顯示,FBXL10(KDM2b)會表達出兩個主要的轉錄本:FBXL10-1是一種較長的轉錄本,這個轉錄本翻譯出來的蛋白在其N端會有組蛋白去甲基化酶結構域,在C端會有F-box、CXXC、PHD、RING和亮氨酸重復結構域;FBXL10-2是一種較短的轉錄本,轉錄起始于另一個外顯子,這個轉錄本翻譯出來的蛋白缺乏組蛋白去甲基化酶結構域,但保留所有其他的結構域。
研究人員還發現,FBXL10-1和FBXL10-2缺失具有明顯不同的表型。FBXL10-2缺失小鼠并不像FBXL10-1缺失小鼠那樣顯示出無腦畸形、缺損或尾巴扭曲的現象。相比之下,FBXL10-2缺失小鼠存活下來的個體具有矮小和顱面發育異常的特征,而產前死亡的個體則顯示顱面和神經管發育異常(圖1)。當與FVB進行回交后,FBXL10∆−2/∆−2胎兒在出生時還有腭裂和眼瞼張開的現象(圖1)[4]。
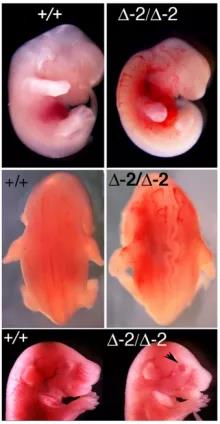
圖1. FBXL10-2等位基因純合缺失的E12.5胚胎(第一行)和E17.5胚胎(第三行)表現出顱面畸形。基因缺失小鼠胎兒頭部前端可以看出明顯受壓的現象,還有出生時睜眼的表型(底部為黑色箭頭)。在Δ-2/Δ-2胚胎中(第二行)能觀察到一個彎曲的神經管[4]。
2●基因敲除小鼠的性別差異:
研究人員通過敲除FBXL10的兩種亞型,構建FBXL10T純合敲除小鼠模型,并使用該模型研究了不同性別之間的表型。與野生型相比,這些胚胎表現出更嚴重的表型和更明顯的性別差異,如圖2所示。胚胎在10.5天就停止發育,并出現多種嚴重發育異常,包括在 Fbxl10∆−2/∆−2敲除小鼠中發現的神經管異常。圖2顯示了由FBXL10-1和FBXL10-2基因敲除介導的性別差異;我們可以觀察到,發育程度最低的雌性胚胎與發育程度最高的雄性胚胎在大致相同的階段停止發育[4]。
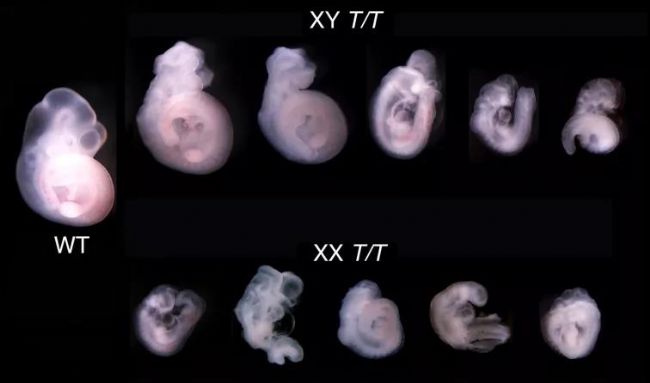
圖2. Fbxl10T/T胚胎的性別差異表型解剖圖。胚胎收集于胚胎期10.5天,通過Sry基因鑒定性別[4]。
3●Fbxl10缺陷型胚胎干細胞仍保留有干性
研究人員從囊胚中分離獲得Fbxl10T等位基因純合的ES細胞,并在培養基中擴增。研究人員發現,突變ES能表達多能性標記物NANOG和OCT4(圖3a),并且能夠嵌合在野生型胚胎中,并支持所有組織生長分化(圖3b)。DNA甲基轉移酶基因表達的分析進一步證明Fbxl10T/TES細胞具有未分化的ES細胞的特征,Fbxl10T/T細胞表達未分化ES細胞和配子發生特定階段的生殖細胞中特有的Dnmt3a(Dnmt3A2)和Dnmt3L。除此之外,Dnmt1和Dnmt3B在野生型ES和在Fbxl10T/TES中的表達水平也非常相似。
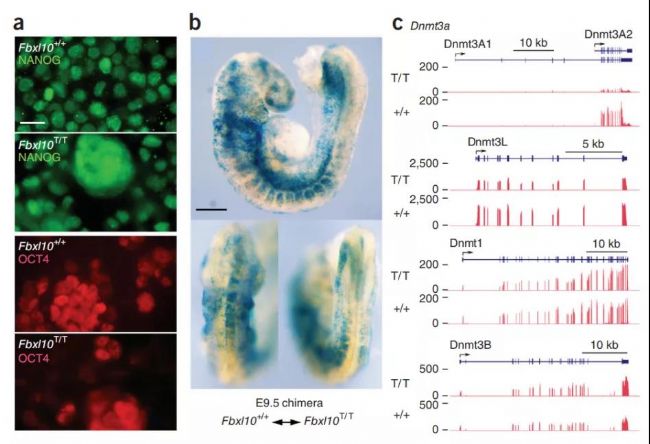
圖3. Fbxl10T/T胚胎干細胞保留有多能性和胚胎干細胞特征。(a) 多能性標記物NANOG(左上)和OCT4(左下)的表達。(b) 注射到WT囊胚后,突變ES細胞對E9.5胚胎所有組織的發育提供了支持,表現出了突變ES細胞的多能性。(c) DNA甲基轉移酶表達譜分析。ES細胞特異性Dnmt3A2和Dnmt3L在野生型和Fbxl10T/T ES細胞中均有表達;Dnmt3L在分化細胞中不表達。除此之外,Dnmt1和Dnmt3B在突變型和野生型ES細胞中的表達譜是一致的[5]。
4●Fbxl10基因異常會導致前腦畸形
有研究發現Fbxl10+/−雜合胚胎表現出中腦和后腦神經管未閉合,以及前腦畸形的特征。所有前腦異常的Fbxl10−/−小鼠在出生后不久都死于中樞神經系統紊亂,除此之外,在E18.5時,小鼠還表現出腦出血和體型較小(圖4G和H)。此外,Fbxl10−/−的胚胎還表現出眼組織缺損和尾巴卷曲的特征(圖4F),異常率分別為40%和10%[6]。
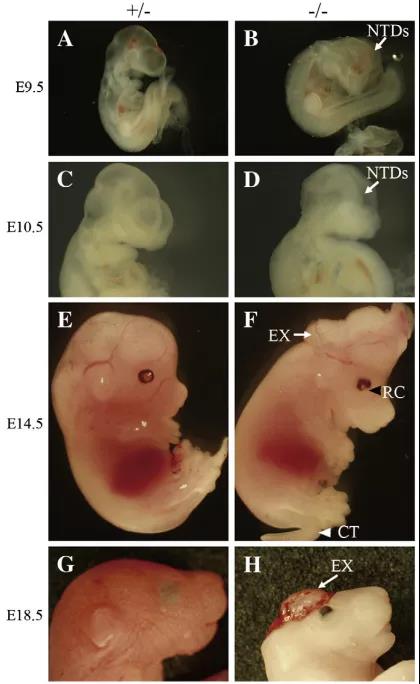
圖4. Fbxl1缺陷的胚胎表型。(A–H)Fbxl10-/-和對照組不同階段的胚胎的側視圖[6]。
A–D:胚胎期9.5天和10.5天胚胎側視圖,對照組胚胎(A和C)的神經管已經關閉,而突變型胚胎中的神經皺襞仍然沒有閉合(B和D,箭頭)。
E和F:在胚胎期14.5天中,一些Fbxl10-/-胚胎(F)的表型,包括中前腦發育異常(E)(箭頭)、眼組織缺損(黑色三角)和尾部卷曲(白色三角)。
G和H:在胚胎期18.5天,Fbxl10-/- 胚胎(H)比對照組胚胎(G)小,并且由于前腦出血而全身蒼白。
小結
KDM2B作為一個能調控染色質組織結構以及組蛋白表觀遺傳修飾的重要蛋白,研究人員對其研究也越來越深入。KDM2B對于神經及腦的發育具有重要的作用,未來的研究可能將集中于該蛋白如何在分子層面調控模式,以及該蛋白對于染色質組織和組蛋白修飾方面的影響。
賽業“紅鼠資源庫”涵蓋基因敲除小鼠、條件性敲除小鼠及海量突變位點小鼠品系,強大的數據庫給您更便捷的體驗,研究人員能夠在線查詢、設計和優化基因編輯方案并比較研究數據和成果,并咨詢訂購。
參考文獻:
1. Boulard M , Edwards J R , Bestor T H . Abnormal X chromosome inactivation and sex-specific gene dysregulation after ablation of FBXL10[J]. Epigenetics & Chromatin, 2016, 9(1):1-9.
2. Boulard M , Edwards J R , Bestor T H . FBXL10 protects Polycomb-bound genes from hypermethylation[J]. Nature Genetics, 2015, 47(5):479-85.
3. Fukuda T , Tokunaga A , Sakamoto R , et al. Fbxl10/Kdm2b deficiency accelerates neural progenitor cell death and leads to exencephaly.[J]. Molecular & Cellular Neuroscience, 2011, 46(3):614-624.



