
ҢУОцМоБПүЫГьСРҫҝөД·ЁТҺ(guЁ©)ТӘЗуЎўУ°н‘ТтЛШј°СРҫҝІЯВФ
ҢУОц№ӨЛҮТСИХқuіЙһйЙъОпЦЖЖ·Йъ®a(chЁЈn)№ӨЛҮЦРІ»ҝЙ»тИұөД№ӨЛҮІҪуEЎЈНЁіЈЈ¬Т»ӮҖЙъ®a(chЁЈn)№ӨЛҮРиТӘФO(shЁЁ)УӢ¶аӮҖХэҪ»өДҢУОцІҪуEҒнЯ_өҪЭ^ёЯөДДҝҳЛјғ¶ИЛ®ЖҪЈ¬ЧоҙуПЮ¶ИөШңpЙЩҡҲБфлsЩ|(zhЁ¬)Л®ЖҪЈ¬ІўқMЧгЕcНвФҙРФОпЩ|(zhЁ¬)УРкP(guЁЎn)өД°ІИ«ҳЛңКЎЈ
№ӨЛҮөДІЩЧчіЙұҫәЬҙуіМ¶ИЙПКЗУЙҢУОцМоБПөДіЙұҫтҢ(qЁұ)„УөДЈ¬ТтҙЛҢУОцМоБПҝЙК№УГөДЕъҙО»тСӯӯh(huЁўn)ҙО”ө(shЁҙ)ЈЁҢУОцМоБПүЫГьЈ©ҢҰЙъ®a(chЁЈn)іЙұҫөДҝШЦЖЦБкP(guЁЎn)ЦШТӘЎЈ
ёчҮшұO(jiЁЎn)№ЬІҝйTіцУЪҢҰЛҺЖ·°ІИ«РФәНУРР§РФөДҝјБҝЈ¬Жд·ЁТҺ(guЁ©)ОДјюГчҙ_ТҺ(guЁ©)¶ЁБЛҢҰ“ҢУОцМоБПөДК№УГүЫГьј°ҝЙҪУКЬПЮ¶И”өДТӘЗуЎЈ
Table 1. ҢУОцМоБПүЫГьСРҫҝөД·ЁТҺ(guЁ©)ЦёДПТӘЗу

ҙЛНвЈ¬2021ДкЈ¬PDA°l(fЁЎ)ІјБЛјјРg(shЁҙ)Ҳуёж60-3Ў¶№ӨЛҮтһЧCЈәЙъГьЦЬЖЪ·Ҫ·Ё ёҪјю2ЈәЙъОпЦЖЛҺФӯБПЛҺЙъ®a(chЁЈn)Ў·Ј¬ҲуёжЦРГчҙ_МбіцМбёЯҢУОцҪйЩ|(zhЁ¬)өДПакP(guЁЎn)тһЧCКЗ№ӨЛҮтһЧCөДЦШТӘІҝ·ЦЎЈ
ҢУОцМоБПФЪК№УГЯ^іМЦРЈ¬•юУРәЬ¶аУ°н‘ЖдүЫГьөДТтЛШЈ¬ЦчТӘ°ьАЁТФПВҺЧьcЈә
ЈЁ1Ј© Ф“ІҪуEФЪјғ»Ҝ№ӨЛҮЦРөДО»ЦГәНҒнБПөДРФЩ|(zhЁ¬)ЎЈЈЁТ»°г¶шСФЈ¬І¶«@ҙЦјғлA¶ОМоБПүЫГьЛҘңpөГёьҝмЈ¬ҫ«јғлA¶ОМоБПүЫГьЛҘңpөГВэЈ©
ЈЁ2Ј© ҢУОцМоБПөДФЩЙъЎўЗеқҚәНұЈҙжіМРтЎЈ
ЈЁ3Ј© ҢУОцЦщСbМоәН“pәДЎЈ
ҢУОцМоБПүЫГьСРҫҝІЯВФұШнҡДЬүтНкИ«·ҙУіЙМҳI(yЁЁ)»ҜЙъ®a(chЁЈn)ТҺ(guЁ©)ДЈөДРФДЬЈ¬»тХЯұШнҡДЬуw¬F(xiЁӨn)ЕcЙМҳI(yЁЁ)»ҜЙъ®a(chЁЈn)ТҺ(guЁ©)ДЈЦ®йgөДПакP(guЁЎn)РФЎЈ
Table 2. ҢУОцМоБПүЫГьСРҫҝІЯВФ

ЧўЈәұO(jiЁЎn)ҝШоlВКЈЁNЈ©ҝЙёщ“ю(jЁҙ)ҢҰ№ӨЛҮөДАнҪвәНҢҚлHЗйӣrЧцЯm‘ӘРФХ{(diЁӨo)ХыЎЈ
• ҝsРЎДЈРНЈЁScale DownЈ©үЫГьтһЧCЗ°Х°РФСРҫҝРиТӘК№УГҝsРЎДЈРНҒнЯMРРЎЈФ“ҝsРЎДЈРНІЙУГҫҖРФҝsРЎөДФӯ„tЈ¬јҙҫSіЦЦщҙІёЯ¶ИЎўҫҖРФБчЛЩЎўЙПҳУЭdБҝЈЁҝЙІЙУГЧоІо—lјюЈ©ЎўҫҸӣ_ТәУГБҝЈЁCVЈ©әНПҙГ“КХјҜҳЛңКІ»ЧғЈ¬ҝsРЎҢУОцЦщөДЦщЦұҸҪЎўуw·eБчЛЩәНҳУЖ·уw·eЈ¬Н¬•rкP(guЁЎn)ЧўҢУОцЦщүәБҰЎўҳУЖ·”UЙўЎўПөҪy(tЁҜng)ЛАуw·eәНЦщР§өИТтЛШЎЈ
Т»°гҝЙК№УГ№ӨЛҮұнХчЖЪйgҪЁБўәНҙ_ХJөДҝsРЎДЈРНЈ¬ТтһйТСҪӣ(jЁ©ng)ҙ_ХJБЛЛьҝЙТФҙъұнЙМҳI(yЁЁ)»ҜТҺ(guЁ©)ДЈөД№ӨЛҮЎЈ
• ҝХ°ЧЯ\РРЈЁCarryoverЈ©ҢУОцМоБПүЫГьСРҫҝөДҢҚтһФO(shЁЁ)УӢРиТӘҝј‘]ҝХ°ЧЯ\РРЈЁCarryoverЈ©ЎЈЦЬЖЪРФөШЯMРРҝХ°ЧЯ\РРҝЙТФУГУЪФu№АЗеқҚіМРтР§№ыәН®a(chЁЈn)Ж·ҡҲБфіМ¶ИЎЈФЪҝХ°ЧЯ\РРЦРЈ¬ҢУОцЦщ°ҙХХіЈТҺ(guЁ©)өДІҪуEІЩЧчЈ¬І»Н¬Ц®МҺФЪУЪЈ¬УГЛ®»тХЯҫҸӣ_ТәҙъМжҒнБПЙПҳУЎЈ
ҢУОцҪйЩ|(zhЁ¬)өДК№УГүЫГьёщ“ю(jЁҙ)ҷzңyЦёҳЛөДҳЛңКҒн¶ЁБxЎЈҷzңyЦёҳЛөДҳЛңКҝЙТФФO(shЁЁ)¶ЁһйТ»ӮҖҫЯуwөДҪШЦ№ҳЛңКЈ¬ұИИзКХВКЈҫ80%Ј¬SEC-HPLC јғ¶ИЈҫ95%өИЈ¬ТІҝЙТФНЁЯ^ҷzңyСӯӯh(huЁўn)ҙО”ө(shЁҙ)йgөДТ»ЦВРФҒн¶ЁБxЈ¬ҙЛ•rҫНІ»ФЩФO(shЁЁ)ЦГҫЯуw”ө(shЁҙ)ЦөҳЛңКЎЈ
Table 3. үЫГьСРҫҝөДФuғrЦёҳЛј°ЖдҝЙҪУКЬҳЛңКЈЁЕeАэЈ©

ҢУОцМоБПүЫГьСРҫҝөДҪKьcЕР¶ЁҝЙТФЯx“сЯ_өҪҷzңyҳЛңКөДСӯӯh(huЁўn)ҙО”ө(shЁҙ)”ө(shЁҙ)ЦөЈ¬»тХЯіц¬F(xiЁӨn)Еъйg®җіЈөДСӯӯh(huЁўn)ҙО”ө(shЁҙ)”ө(shЁҙ)ЦөЎЈНЁіЈЭ^һйұЈКШөДЧц·ЁКЗҢўҙ_¶ЁөДСӯӯh(huЁўn)ҙО”ө(shЁҙ)”ө(shЁҙ)ЦөіЛТФТ»ӮҖ°ІИ«Пө”ө(shЁҙ)Ј¬Ғн¶ЁБxЧоҪKөДҢУОцМоБПүЫГьҙО”ө(shЁҙ)ЎЈ
Novo-A DiamondУHәНМоБПКЗІ©ёсВЎЧФЦчСР°l(fЁЎ)өДРВТ»ҙъёЯДНүAProtein Aҝ№уwУHәНМоБПЈ¬ҝЙДНКЬ0.5M~1.0M NaOHЯMРРCIPЈ¬МоБПөДЭdБҝёьёЯЎўүәБҰБчЛЩРФДЬёьәГЎЈ
ұҫ°ёАэК№УГә¬УРҶОҝЛВЎҝ№уwөДјҡ°ыЕарB(yЁЈng)ЙПЗеЈ¬ҢҰNovo-A DiamondУHәНМоБПЯMРРБЛ188ӮҖСӯӯh(huЁўn)өДңyФҮЈ¬УГ0.1M NaOHәН0.5M NaOHЯMРРCIPЈ¬Н¬•rҷzңyПҙГ“КХВКЎўHCPЎўHCDЎўProtein AЎўSECЦёҳЛЎЈ
ңyФҮ”ө(shЁҙ)“ю(jЁҙ)ұнГчNovo-A DiamondҫЯУРЭ^ёЯөДЭdБҝЈЁіхКј10%DBCһй67 mg/mLЈ©Ј¬Я\РР156ӮҖСӯүДәуТАИ»ёЯУЪіхКј10%DBCөД80%Ј»ЛщУРСӯӯh(huЁўn)өДКХВК·Җ(wЁ§n)¶ЁЈ¬ПҙГ“ҳУЖ·јғ¶И·Җ(wЁ§n)¶ЁЈ¬HCPЎўHCDәНө°°ЧAөДҡҲБфКјҪKМҺУЪЭ^өНөДЛ®ЖҪЎЈ
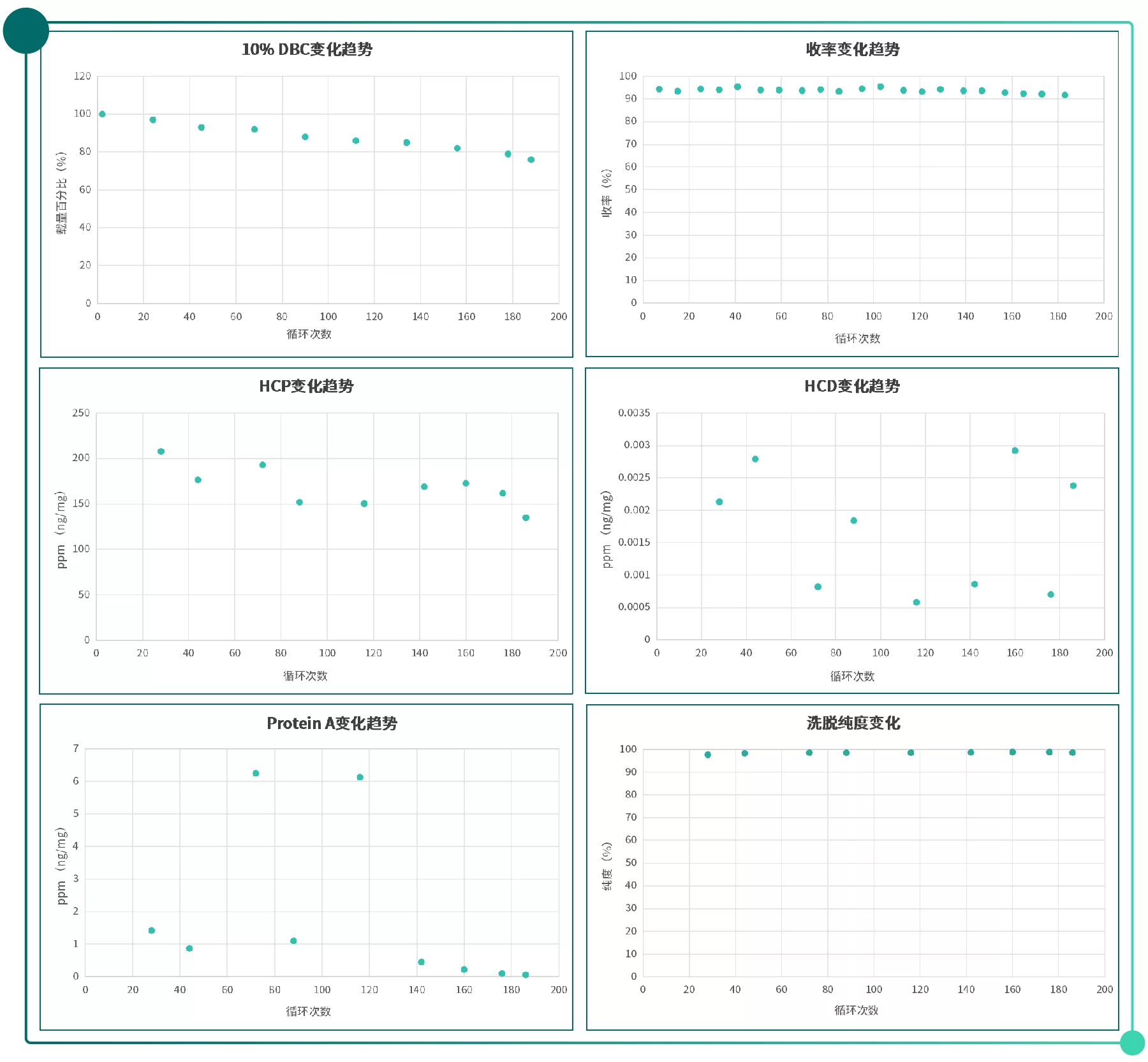
Fig.1 Novo-A Diamond үЫГьСРҫҝёчн—”ө(shЁҙ)“ю(jЁҙ)ҷzңyҪY(jiЁҰ)№ыҲDЧV
ҢУОцМоБПЧчһйЙъОпЦЖЛҺПВУОјғ»Ҝ№ӨЛҮЦРЦБкP(guЁЎn)ЦШТӘөДФӯІДБПЈ¬ЖдРФДЬЕcЙъОпЛҺОпөД®a(chЁЈn)Ж·Щ|(zhЁ¬)БҝГЬЗРПакP(guЁЎn)ЎЈНЁЯ^МоБПүЫГьСРҫҝҢҚтһЈ¬І»ғHғHМбёЯБЛЙъ®a(chЁЈn)№ӨЛҮөДҝЙҝШРФЕcҪӣ(jЁ©ng)қъРФЈ¬ТІһйЙМҳI(yЁЁ)»ҜЙъ®a(chЁЈn)МṩҝЙҝҝұЈЧCЎЈ
…ўҝјЩYБП
[1] 2020 °жЎ¶ЦРҮшЛҺөдЎ·ИэІҝ“ИЛУГЦШҪMҶОҝЛВЎҝ№уwЦЖЖ·ҝӮХ“”.
[2] FDA “Guidance for IndustryЈәProcess Validation: General Principles and Practices”.
[3] FDA “Points to Consider in the Manufacture and Testing ofMonoclonal Antibody Products for Human Use”.
[4] EMA “Guideline on process validation for the manufacture of biotechnology-derived active substances and data to be provided in the regulatory submission”.
[5] EMA “CPMP Position Statement on DNA and Host Cell Proteins (HCP) Impurities, Routine Testing Versus Validation Studies,” CPMP/BWP/382/97.
[6] PDA Technical Report No.60-3: Process validation: A lifecycle approach Annex 2: biopharmaceutical drug substances manufacturing. Parenteral Drug Association: 2021.
В“(liЁўn)ПөлҠФ’Јә400-820-5172
E-mailЈәinfo@bestchrom.com

- Ҹ—РФуwөДІДБП»ҜҢWМШРФЎў·NоҗЯx“сј°ҫCәП‘ӘУГ
- ҢУОцМоБПүЫГьСРҫҝөД·ЁТҺ(guЁ©)ТӘЗуЎўУ°н‘ТтЛШј°СРҫҝІЯВФ
- ҢУОцјјРg(shЁҙ)өДЛДҙуҪӣ(jЁ©ng)өд·ҪКҪј°Жд·ЦлxФӯАнЎўғһ(yЁӯu)„ЭЎўЯmУГҲцҫ°әНЯx“сТӘьc
- ОД«I·ЦПнЈәProtein AҢУОцёДЯMРВІЯВФЦъБҰҪвӣQҝ№уwҫЫјҜуwИҘіэлyо}
- ИзәОМбЙэлpҝ№јғ»ҜР§№ыЈәТ»ОДЧx¶®pHМЭ¶ИПҙГ“өДғһ(yЁӯu)„ЭЕcМф‘р(zhЁӨn)
- ЙъОпЦЖЖ·ҝЙұИРФСРҫҝөДТвБxЎўСРҫҝғИ(nЁЁi)ИЭәНСРҫҝҪY(jiЁҰ)№ы
- ОД«I·ЦПнЈәPEGЕcҫ«°ұЛбЎӘЎӘҝ№уwјғ»ҜЦРИҘіэҫЫјҜуwөДөГБҰЦъКЦ
- СӘТәЦЖЖ·ЈЁBlood ProductsЈ©јғ»ҜіЈУГөД·Ҫ°ё·ЦПн
- ПајsіЙ¶јЈәІ©ёсВЎСыДъ…ўјУ2025ОҙҒнXDCРВЛҺҙу•ю
- І©ёсВЎҫ«ІКББПаBIOCHINA2025өЪК®ҢГТЧЩQ(mЁӨo)ЙъОп®a(chЁЈn)ҳI(yЁЁ)ҙу•ю
- ПајsМKЦЭЈәІ©ёсВЎСыДъ…ўјУBIOCHINA2025
- І©ёсВЎПsВ“(liЁўn)2024ЦРҮшЙъГьҝЖҢW·ю„ХЖуҳI(yЁЁ)Ж·ЕЖ100ҸҠ
- СРУ‘•юЈәҝ№уwоҗЛҺОпПВУОјғ»Ҝ№ӨЛҮкP(guЁЎn)жIТӘЛШҪвОц
- І©ёсВЎ7ҝоГчРЗҢУОцМоБП®a(chЁЈn)Ж·ПЮ•rГвЩMФҮУГЈ¬ПИөҪПИөГ
- І©ёсВЎНЖіцҝ№уwјғ»ҜРВЖ·AT Protein A Diamond Ultra
- І©ёсВЎ№ЩҫW(wЁЈng)ҹЁРВЙПҫҖЈ¬·ю„ХуwтһИ«ГжЙэјү


