
一小時蛋白質組: 基于Q-OT-qIT質譜系統的蛋白質組學快速鑒定
張偉†, 顧培明†, 江崢*, 明紅, 陳偉
賽默飛世爾科技(中國)有限公司, 上海 201206
† 同等貢獻
* 聯系人, E-mail: zheng。jiang@thermofisher。com
摘要 隨著“一小時酵母蛋白質組”的實現, 短梯度下實現蛋白質組深度覆蓋成為可能。本文利用全新Q-OT-qIT三合一質譜系統進行“一小時蛋白質組”分析與優化。50 min有效梯度, 單次實驗分別從1 μg和50 ng HeLa全蛋白中鑒定到20860和14100條肽段, 對應到3865和2877個非冗余蛋白, 而目前的文獻報道至少需要2~3 h。同時, 本文考察了Q-OT-qIT碎裂模式、檢測方法、最大注入時間和自動增益控制等參數對蛋白質組快速分析的影響, 證明了不同采集方法間的互補性, 闡述了不同掃描參數對鑒定結果的影響。此外, 還討論了Q-OT-qIT的并列運行原理和掃描組合模式, 為不同實驗目的和樣本類型的蛋白質組快速分析奠定了基礎。
關鍵詞 蛋白質組學 快速鑒定 深度覆蓋 靜電場軌道阱 Q-OT-qIT質譜
蛋白質組深度覆蓋分析(in-depth proteome)已成為蛋白質組學研究的發展趨勢, 通過大規模鑒定和挖掘極低豐度的蛋白, 為信號通路、分子靶點和生物標志物的研究提供重要信息。然而, 由于全蛋白樣本的復雜性, 蛋白質組深度鑒定對色譜分離要求很高, 通常需要進行二維分離(強陰離子交換、高pH反相等), 或使用一維長梯度分離, 以降低高豐度蛋白的干擾[1,2]。但這樣消耗了大量時間, 重現性也不佳, 成為蛋白質組深度覆蓋研究的瓶頸。
靜電場軌道阱(orbitrap)質量分析器具有超高分辨率、超高靈敏度等特點, 有效推動了蛋白質組學的發展[3,4]。新型Q-OT-qIT(Orbitrap Fusion)質譜系統首 次將四極桿(quadrupole, Q)、靜電場軌道阱(orbitrap, OT)和線性離子阱(linear quadrupole ion trap, qIT)3種質量分析器集為一體, 利用動態掃描管理技術, 實現了3種質量分析器同時工作并相互協作, 一級、二級掃描同時進行, 使分析效率達到最大化[5]。Hebert等人[6]利用Q-OT-qIT質譜, 5次重復實驗, 在70min有效梯度下鑒定到約4000個酵母蛋白, 同樣的結果, 目前的文獻報道至少需要4h, 分析效率(單位時間蛋白鑒定數量)提高了4倍以上[7,8]。基于此,Hebert等人[6]首次提出“一小時酵母蛋白質組”的概念, 即1h左右的短梯度實現酵母蛋白質組深度覆蓋。蛋白質組學已進入新的時代。
本文使用Q-OT-qIT質譜系統對HeLa和酵母細胞全蛋白進行短梯度快速鑒定, 并對基于Q-OT-qIT質譜的“一小時蛋白質組”深入考察與優化。結果顯示, 單次實驗、50min有效梯度, 分別從1 μg和50 ng HeLa中鑒定到20860和14100條肽段, 對應3865和2877個非冗余蛋白, 分析效率相比目前的報道提高了至少2~3倍[9]。本文還對比和優化了多種掃描組合和參數設置, 為各類實驗提供了最合適的采集模式。
1 材料與方法
1.1 樣品前處理
分別取適量HeLa和酵母細胞沉淀, 加入含7 mol/L尿素、2 mol/L硫脲、1 mmol/L苯甲基磺酰氟(phenylmethanesulfonyl fluoride,PMSF)和50 mmol/L二硫蘇糖醇(DL-dithiothreitol,DTT)的蛋白提取液(使用前加入蛋白酶抑制劑), 在超聲細胞破碎儀中超聲3×5s. 冰上放置20min后, 于4℃離心30min(15000×g)并取上清, Bradford法測定總蛋白濃度.
向蛋白提取液中加入DTT, 使終濃度為10 mmol/L, 于56℃振蕩30min. 再加入碘乙酰胺(iodoacetamide, IAM), 使終濃度為50 mmol/L, 于室溫避光振蕩40min. 反應后, 加入6倍體積的預冷丙酮, 20℃放置3h, 再于4℃離心30min (15000×g)棄上清. 按10 μg/μL的濃度將沉淀重溶于50 mmol/LNH4HCO3緩沖溶液, 并加入足量胰蛋白酶(trypsin), 37℃酶解過夜. 酶解后, 將全蛋白酶解液稀釋至終濃度為0.5 μg/μL.
1.2 納流液相色譜方法
色譜儀: 納流速超高效液相色譜EASY-nLC 1000(Thermo, 美國); 色譜柱: 納流C18EASY-Spray柱(2 μm, 100 Å, 75 μm×25 cm/50 cm), 流速250nL/min; 自制納流C18柱(3 μm, 100 Å, 75 μm×15 cm), 流速300nL/min; 流動相A: 0.1%甲酸-水溶液;流動相B: 0.1%甲酸-乙腈溶液.
50min有效梯度: 0~2min, 3%~5% B; 2~42min, 5%~22% B; 42~52min, 22%~30% B; 52~55min, 30%~90% B; 55~60min, 90% B. 70min有效梯度: 0~5 min, 3%~8% B; 5~60min, 8%~20% B; 60~75min, 20%~30% B; 75~80min, 30%~90% B; 80~90min, 90% B.
1.3 質譜方法
質譜儀: Orbitrap Fusion(Q-OT-qIT)高分辨質譜儀(ThermoFisher, 美國); 離子源: EASY-Spray或Nanospray Flex納升電噴霧源; 噴霧電壓: 2.2 kV; 離子傳輸管溫度: 275℃; RF-lens: 60%.
掃描模式: Top-speed; 循環時間: 3 s; 動態排除: 60 s.
一級掃描: 質量分析器,軌道阱; 掃描范圍,m/z350~1800; 分辨率,120k; 最大注入時間,50 ms; 自動增益控制(AGC): 2e5.
二級IT掃描: 離子選擇,四級桿; 選擇窗口,2 amu; 質量分析器,離子阱(ion trap, IT); 掃描速度,Rapid; 最大注入時間,35 ms; AGC: 1e4; 碎裂模式,HCD; 碎裂能量, 35%.
二級OT掃描: 離子選擇,四級桿; 選擇窗口,2 amu; 質量分析器,軌道阱; 分辨率,15k; 最大注入時間,35 ms; AGC,1e5; 碎裂模式,HCD; 碎裂能量, 35%.
1.4 數據分析
數據使用Proteome Discoverer 1.4軟件搜庫鑒定. 搜索引擎: SEQUEST HT; 數據庫: Uniprot人和酵母蛋白數據庫; 蛋白酶: 胰蛋白酶(full); 最大漏切位點: 2; 母離子質量精度: 10 ppm; 子離子質量精度: 0.6 Da(qIT)/0.02 Da(OT); 固定修飾: Carbamidomethyl(C+57.021 Da); 可變修飾: acetyl(N-terminus+42.011 Da), deamidated(N, Q+0.984 Da), oxidation(M+15.995 Da). 搜庫結果使用Percolator計算q值進行卡值. 同時, 使用Preview軟件分析數據質量.
2 結果與分析
2.1 Q-OT-qIT短梯度分析HeLa和酵母蛋白質組
實驗對1 μg和50 ng HeLa進行50min有效梯度采集, 對1 μg酵母全蛋白進行70min有效梯度采集, 采用OT-HCD-IT掃描模式, 一級軌道阱、二級離子阱掃描, HCD高能碎裂. 結果顯示, 有效出峰時間蛋白酶解產物得到充分分離, 質譜響應良好(圖1). 即使在50 ng的極低樣本量下, 仍獲得良好的分離效果和質譜響應(圖1B).
1 μg HeLa樣本在50min有效梯度內共獲得53209張譜圖, 平均每秒1.7張一級譜圖、16.1張二級譜圖, 平均每循環9.6張二級譜圖; 酵母樣本在70min有效梯度內共獲得60295張譜圖, 平均每秒1.8張一級譜圖、12.5張二級譜圖, 平均每循環6.9張二級譜圖(表1). HeLa細胞比酵母復雜, 存在更多的蛋白, 因此獲得更多的二級譜圖. 此外, 在50 ng HeLa的極低樣本量下, 總譜圖數也達到41099張.


圖1 HeLa和酵母蛋白酶解產物的LC-MS/MS總離子流圖
A: 1 μg HeLa; B: 50 ng HeLa; C: 1 μg酵母
表1 有效梯度內的采集數據分析
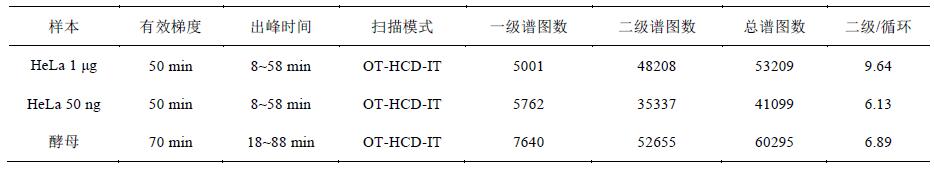
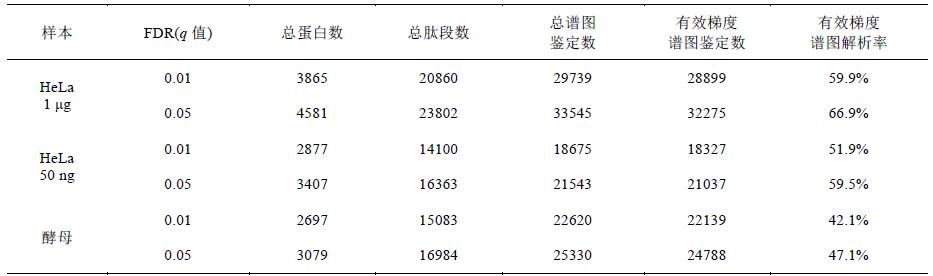
表2 HeLa和酵母樣本全蛋白鑒定結果
2.2 HeLa和酵母蛋白質組快速鑒定結果
數據經SEQUEST HT搜庫和Percolator卡值, 從1 μg和50 ng HeLa樣本中分別鑒定到20860和14100條肽段(q值0.01), 分別對應3865和2877個非冗余蛋白(表2), 而同樣的鑒定數量, 目前的報道至少需要2~3h[9]. 另外, 酵母樣本共鑒定到15083條肽段(q值0.01), 對應2697個非冗余蛋白(表2).
HeLa數據進一步使用Preview軟件分析譜圖質量精度(圖2). 結果顯示, 母離子(m/z)質量精度平均為0 ppm, 子離子質量精度(m/z)平均為0.075 Da. 軌道肼質量偏差基本保持在5 ppm以內, 具有高質量精度和長期穩定的質量軸; 離子阱質量偏差基本保持在±0.2 Da以內, 質量精度良好, 保證了數據的質量與結果可信度.
肽段ATAGDTHLGGEDFDNR的譜圖展示了Q-OT-qIT質譜的碎裂效果和靈敏度(圖3), 譜圖具有豐富的碎片信息、良好的匹配度和較高的靈敏度. 此外, HCD和CID兩種碎裂模式效果相當并有著各自的特點: HCD避免了CID的三分之一效應, 低分子量端信息沒有丟失, 同時速度比CID快5%; 而CID靈敏度更高, 對低豐度肽段碎裂效果更明顯.
2.3 不同碎裂模式和檢測方法比較與分析
鑒于不同模式的鑒定效果差異明顯, 實驗深入考察了HCD/CID碎裂模式和軌道阱/離子阱二級檢測方法對鑒定結果的影響. 首先分析了HeLa在OT-HCD-IT, OT-CID-IT和OT-HCD-OT3種模式下的鑒定結果, 比較其互補性. 結果表明, HCD和CID共同鑒定到的肽段占32.7%, 僅一種碎裂模式鑒定到的占67.3%; OT/IT同時檢測到的肽段占51.9%, 僅一種質量分析器檢測到的占48.1%(圖4). CID有三分之一效應, 造成低分子量端的質量歧視, HCD克服了低質量端歧視, 但靈敏度略低于CID; 離子阱具有較高的掃描速度和靈敏度, 而軌道阱的高分辨率與高質量精度有效排除了復雜樣本基質的干擾. 因此, 兩種碎裂模式和檢測方法具有一定的互補性.
實驗進一步使用OT-HCD-IT, OT-CID-IT, OT-HCD-OT和OT-CID-OT4種掃描模式, 對酵母樣本進行60 min有效梯度采集, 詳細比較了不同掃描模式的差異. 其中, 二級IT采用Rapid掃描, OT采用30k分辨率. 鑒定結果以OT-HCD-IT結果為100%歸一化(圖5). 結果表明, 由于OT-HCD-IT速度最快, 獲得的譜圖數和鑒定結果最多; CID比HCD慢5%, 因此鑒定結果少5%; OT-HCD-OT為兩級高分辨, 一級、二級無法同時運行, 譜圖數和蛋白鑒定數分別是二級IT掃描的70%和85%; OT-CID-OT速度最慢, 譜圖數和蛋白鑒定數分別是OT-HCD-IT的60%和75%. 此外, 4種掃描模式的譜圖解析率相差不大, 表明不同掃描模式對數據質量影響不大.
2.4 離子最大注入時間與自動增益控制(AGC)對結果的影響
利用C-trap控制離子的注入時間和注入數量(自動增益控制)是軌道阱質量分析器的特點, 進入軌道阱的離子數量對檢測結果影響很大, 合適的離子數量不僅避免了空間電荷效應, 還能夠明顯提升譜圖質量、動態線性范圍和檢測線. 實驗在OT-HCD-OT模式下設置不同參數對HeLa樣本進行分析(二級OT采用15k分辨率), 進一步探索不同的最大注入時間和AGC設置對結果的影響(表2).
鑒定結果表明, 當樣本濃度較高(1 μg)時, 離子數量充足, 較小的注入時間和AGC即可獲得良好的結果;增加注入時間和AGC反而會降低掃描速度, 減少譜圖數量.因此, 參數設置為1.0e5, 35 ms時獲得的鑒定數最多(圖6A). 當樣本濃度很低(50 ng HeLa)時, 需要增加注入時間和AGC以注入更多的離子, 提高靈敏度和譜圖質量; 但如果注入時間過長(120和200ms), 譜圖數量太少, 也會導致鑒定結果減少.因此, 2.0e5, 60 ms的參數設置獲得的鑒定數最多(圖6B). 此外, 注入時間越長、離子數量越多, 譜圖質量越高, 因此, 離子注入數量與譜圖解析率成正比.
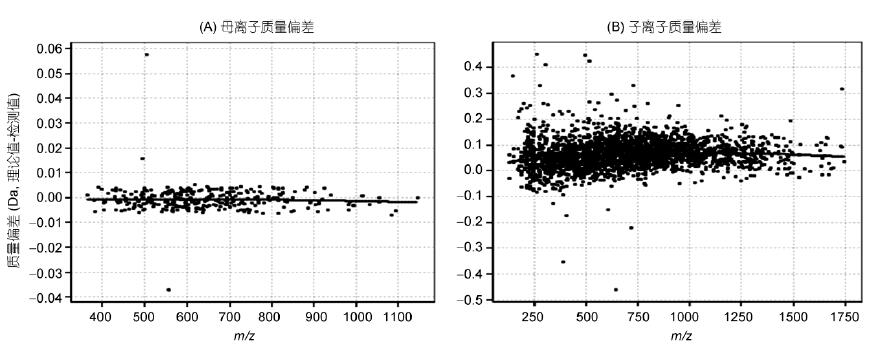
圖2 數據質量精度分析
A: 一級質量精度; B: 二級質量精度
表3 不同的最大注入時間與AGC下HeLa樣本的鑒定結果
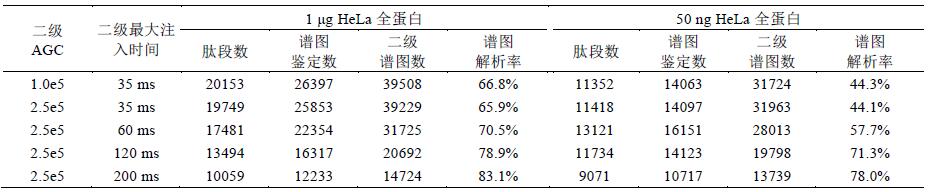
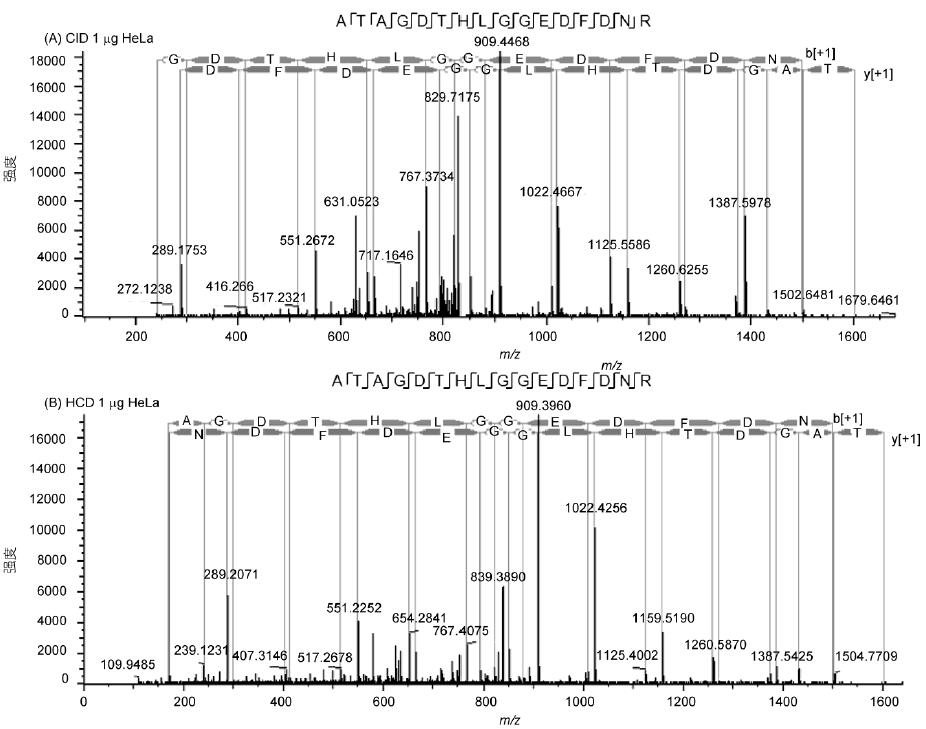
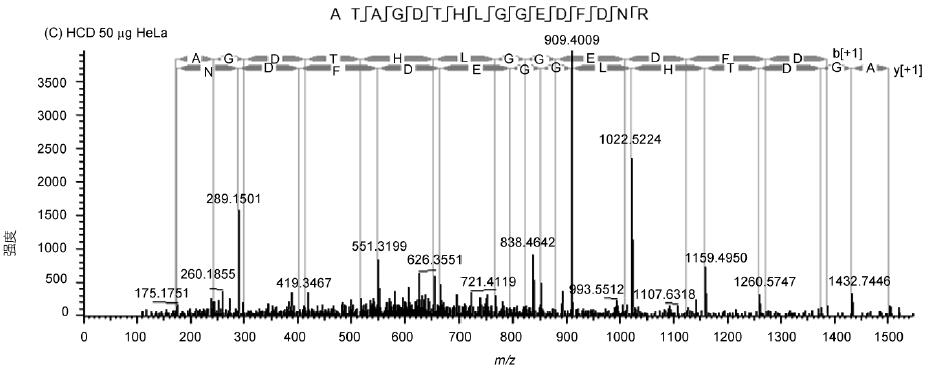
圖3 HeLa樣本ATAGDTHLGGEDFDNR肽段二級譜圖和匹配信息
A: 1 μg樣本量, CID碎裂; B: 1 μg樣本量, HCD碎裂; C: 50 ng樣本量, HCD碎裂
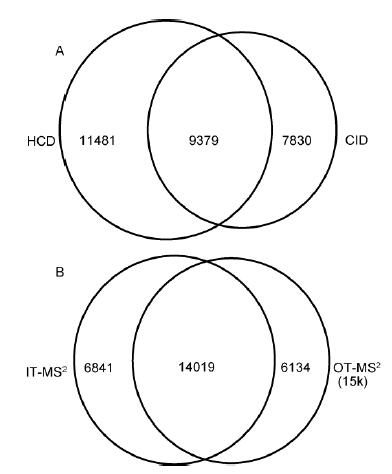
圖4 HeLa樣本肽段鑒定結果比較
A: HCD和CID碎裂模式比較; B: 離子阱和軌道阱二級檢測比較
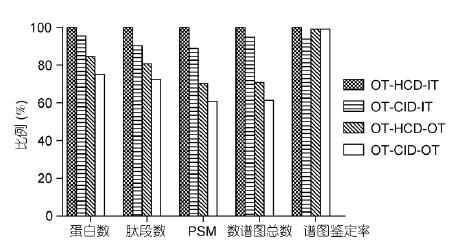
圖5 不同掃描模式數據比較
以OT-HCD-IT為100%歸一化

圖6 不同的最大離子注入時間和AGC結果比較
A: 1 μg HeLa結果比較; B: 50 ng HeLa結果比較. 以每項最高值為100%
3 討論
隨著“一小時酵母蛋白質組”全覆蓋的實現, 蛋白質組深度覆蓋研究迎來新的時代[6]. 在復雜樣本蛋白質組研究中, 傳統方法需要進行二維分離、分級的方式簡化樣本、充分分離, 這樣不僅操作費力, 也需消耗大量的時間. 而Q-OT-qIT質譜技術使得在60 min左右一維短梯度下實現復雜樣本的深度鑒定成為現實.
Q-OT-qIT的三合一結構實現了傳統質譜難以實現的并列運行, 即一級、二級掃描同時進行, 離子注入、選擇、碎裂、檢測同時進行(圖7). 動態掃描管理技術自動控制和實時優化儀器運行, 使3種質量分析器相互配合、同時工作, 盡可能減少掃描過程和循環間隙的等待時間. “減一邏輯”將二級掃描與上一次一級掃描相關聯, 無需等待本次一級掃描確定母離子及電荷, 提高儀器運行速度和分析效率. 以top 10的數據依賴掃描(DDA)為例, 240k分辨率下, 當一級全掃描完成時, 所有二級掃描也同時完成, 一級、二級互不相關、獨立運行(圖7). 此外, top-speed模式在固定時間內盡可能多地激發二級掃描, 相比傳統Top-N模式能更多地采集低豐度肽段, 也保證了一級掃描點數, 有利于同時定性定量.
實驗結果表明, 1 μgHeLa樣本在50 min有效梯度內采集到53209張譜圖, 掃描速度為17.7 Hz, 其中包括120k分辨率的一級軌道阱掃描, 并列運行的工作模式提高了儀器整體運行速度. 在嚴格卡值(1% FDR)下, 鑒定到20860條非冗余肽和3865個非冗余蛋白, 而最新的文獻報道需要至少2~3h才能達到相同結果[9]. 酵母樣本雖未達到Hebert等人[6]報道的蛋白鑒定數量, 但上樣量更低, 并且未進行重復, 比目前其他儀器的報道結果也有2倍以上的提高[8,9]. 在掃描速度方面, Q-OT-qIT雖然未達到Q-TOF類質譜的極限速度, 但譜圖質量更高, 譜圖解析率比Q-TOF類質譜高2~3倍[8].
1 μgHeLa樣本在多次實驗中的譜圖解析率基本達到60%以上, 在加大離子注入時間和注入數量的情況下甚至達到83.1%(表3), 顯示了良好的譜圖質量. 全蛋白樣本體系復雜, 短梯度下會有共流出肽段和基質雜質的干擾, 大量低豐度肽段的二級響應也很弱, 在這樣的情況下, 譜圖解析率達到80%以上, 顯著高于目前的文獻報道[8,9]. 此外, 樣本量決定了蛋白鑒定數量, 傳統蛋白質組學分析需要微克級的全蛋白, 而本實驗嘗試將樣本量降低到納克級, 即50 ng上樣量, 鑒定結果已達到目前微克級的水平, 為低濃度樣本和極低豐度蛋白分析提供可能.
Q-OT-qIT的三合一結構使掃描方式靈活多樣. C-trap與離子阱之間的多極離子通道使離子可以正反向自由移動, 實現了CID/HCD/ETD3種碎裂模式在MSn任意一級使用, 并任意使用軌道阱或離子阱檢測, 產生多種組合的掃描模式. 其中, CID碎裂相比HCD靈敏度略高, 但是速度略慢, 且有三分之一效應, 即低質量端歧視, 而HCD在低分子量段具有豐富的碎片信息; 軌道阱的超高分辨率與質量精度可以最大程度地排除干擾, 而離子阱速度更快, 并且可以和軌道阱同時運行, 提高效率. 可見, 由于機理的不同, 各種掃描模式的效果不同, 具有一定的互補性. Q-OT-qIT控制離子注入時間和注入數量的能力也是保證分析效率和靈敏度的重要因素. 注入時間越長、注入數量越多, 則靈敏度越高、譜圖質量越高, 但掃描速度越慢; 相反, 離子注入數量越少, 速度越快, 但低豐度離子損失越大, 靈敏度下降. 可見, 實驗目的和樣本不同,適合的掃描模式和參數也不同, 因此, 實驗進一步考察了掃描模式和離子注入參數對結果的影響.
結果表明, 不同碎裂模式和檢測方法均具有良好的互補性. 軌道阱和離子阱之間有1/2的鑒定結果不重復, HCD和CID之間有2/3結果不重復, 不同的掃描方法結合可以達到更大的覆蓋范圍. 另一方面, 線性離子阱相比傳統離子阱質量精度更高, 質量偏差穩定保持在0.2 Da以內, 達到中高分辨質譜水平, 因此, OT和IT掃描均具有較高的譜圖解析率. 在離子控制方面, 通過提高最大注入時間和最大注入數量, 可以增加離子注入量, 提高低濃度樣本和極低豐度蛋白的靈敏度; 而降低離子注入量, 能夠加快常規樣本的分析速度、增加鑒定數量. 通過幾種模式和參數的考察、優化, 為蛋白質組快速分析和深度覆蓋研究提供了更多選擇.
4 結論
本文利用Q-OT-qIT三合一質譜, 對HeLa和酵母全蛋白進行了短梯度快速鑒定: (ⅰ) 50 min有效梯度, 從1 μgHeLa中鑒定到20860條非冗余肽和3865個非冗余蛋白, 時間相比目前的報道縮短了50%~60%; (ⅱ) 50 ng極低樣本量, 鑒定到14100條非冗余肽和2877個非冗余蛋白, 譜圖解析率超過50%; (ⅲ) 考察了HCD/CID碎裂模式和IT/OT檢測方法的異同, 結果顯示, 不同方法間互補性良好, 并且均有較高的譜圖解析率; (ⅳ) 考察了離子最大注入時間和自動增益控制對結果的影響, 表明提高注入時間和注入數量可以顯著提高低濃度樣本的檢測靈敏度. 綜上所述, 基于Q-OT-qIT質譜, 本文實現了短時間內對復雜樣本蛋白質組深度覆蓋分析, “一小時蛋白質組”時代已經到來.

圖7 Q-OT-qIT質譜結構與并列運行原理
軌道阱一級全掃描的同時, 四極桿依次進行若干母離子選擇, 離子阱依次進行若干母離子的二級碎片掃描, 三者同時運行
致謝 部分圖片素材由賽默飛世爾科技圣何塞工廠提供.
參考文獻
1 Delahunty C M, Yates III J R. MudPIT: multidimensional protein identification technology. Biotechniques, 2007, 43: 563–569
2 Jones K A, Kim P D, Patel B B, et al. Immunodepletion plasma proteomics by triple TOF 5600 and orbitrap elite/LTQ-orbitrap Velos/Q exactive mass spectrometers. J Proteome Res, 2013, 12: 4351–4365
3 Mann M, Kelleher N L. Precision proteomics: the case for high resolution and high mass accuracy. Proc Natl Acad Sci USA, 2008, 105: 18132–18138
4 Geiger T, Wehner A, Schaab C, etal. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol Cell Proteomics, 2012, 11: M111.014050
5 Senko M W, Remes P M, Canterbury J D, et al. Novel parallelized quadrupole/linear ion trap/orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal Chem, 2013, 85: 11710–11714
6 Hebert A S, Richards A L, Bailey D J, et al. The one hour yeast proteome. Mol Cell Proteomics, 2013: M113.034769
7 Nagaraj N, Kulak N A, Cox J, et al. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top orbitrap. Mol Cell Proteomics, 2012, 11: M111.013722
8 Andrews G L, Simons B L, Young J B, et al. Performance characteristics of a new hybrid quadrupole time-of-flight tandem mass spectrometer (TripleTOF 5600). Anal Chem, 2011, 83: 5442–5446
9 Kelstrup C D, Young C, Lavallee, R, et al. Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. J Proteome Res, 2012, 11: 3487–3497
The One Hour Proteome: Q-OT-qIT Mass Spectrometry Based Rapid Proteome Identification
ZHANG Wei, GU PeiMing, JIANG Zheng, MING Hong & CHEN Wei
ThermoFisher Scientific China, Shanghai 201206, China
With the realization of “the one hour yeast proteome”, in-deep proteome under rapid LC-MS/MS gradient is achieved. The current study utilized novel Q-OT-qIT mass spectrometer to analyze and optimize “the one hour proteome”. Within 50 min LC-MS/MS gradient, a total of 20860 and 14100 unique peptides were identified from 1 μg and 50 ng HeLa digest, corresponding to 3865 and 2877 proteins, respectively. Similar result from current reports used at least 2–3 h gradient. The effect of various collision modes, detectors, maximum injection times and automatic gain control settings were further evaluated, and the complementarity between different acquisition methods and scan parameters were described. Moreover, the massive parallelization of Q-OT-qIT was discussed, providing useful information for rapid proteome analysis.
proteomics, rapid identification, deep coverage, orbitrap, Q-OT-qIT MS
doi: 10.1360/10.1360/052014-16




Copyright(C) 1998-2025 生物器材網 電話:021-64166852;13621656896 E-mail:info@bio-equip.com