
染色質免疫共沉淀測序(ChIP-seq)的數據挖掘思路
CHIP-seq研究的數據挖掘思路主要分為3步:
后期視情況是否需要下游實驗設計驗證TF結合/組蛋白修飾的目標區域和候選靶基因。

1、圖譜分析
(1)peak/reads在基因組上的分布
l Peak的分布就是蛋白與DNA互作圖譜。
l 不同蛋白對DNA的結合可以按照峰的寬窄和分布特征分為: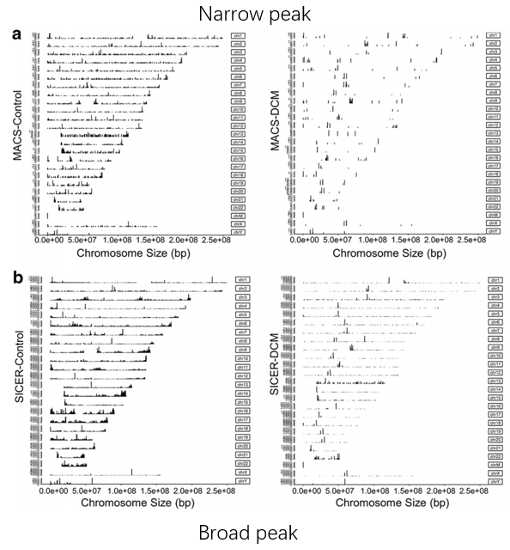
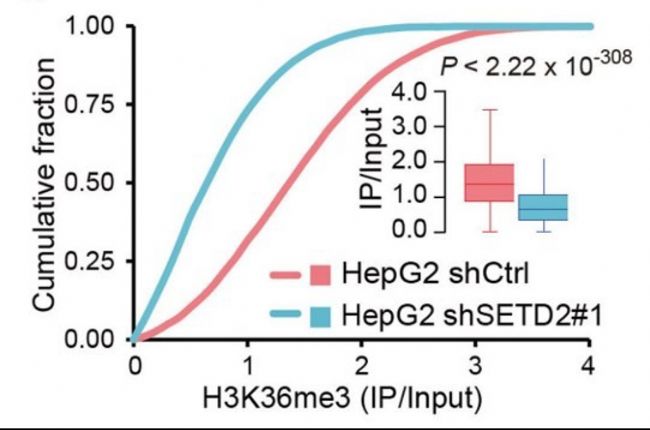
(2)信號的富集程度分析——覆蓋度累積曲線
對樣本比對結果reads累積情況進行展示。一定長度窗口(bin)上reads數進行計數,然后排序,再依次累加畫圖。input (能測到90 DNA片段)在基因組理論上是均勻分布,隨著測序深度增加趨近于直線,實驗組在排序越高的窗口處reads累積速度越快,說明這些區域富集的越特異。
narrow peak :富集程度高;broad peak:富集程度低。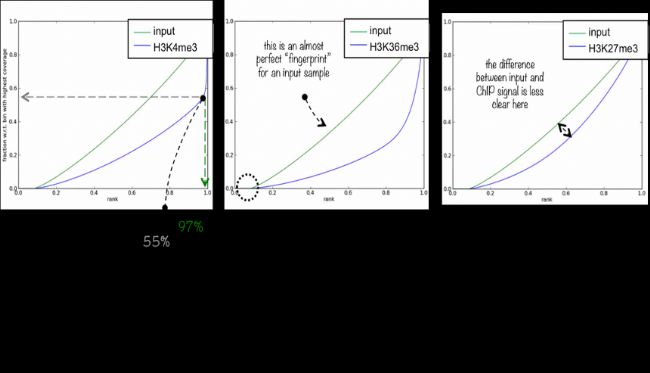
(3)peak/reads的基因元件富集分析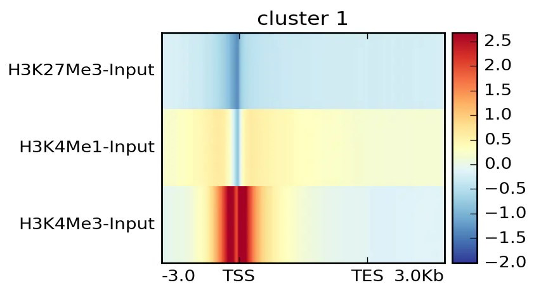
(4)peak/reads的基因元件分布分析
(5)peak/reads與TSS的相對距離分布
轉錄因子、組蛋白修飾往往具有重要的轉錄調控功能,而TSS附近是主要的轉錄調控區域,因此判斷peak與TSS的位置關系有重要的意義。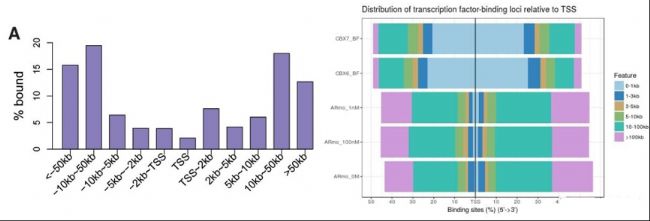
(6)降維分析
將基因組分為等長窗口(bins),計算各樣本各窗口內的Reads覆蓋情況并進行標準化。基于此數據進行相關性、聚類和PCA分析。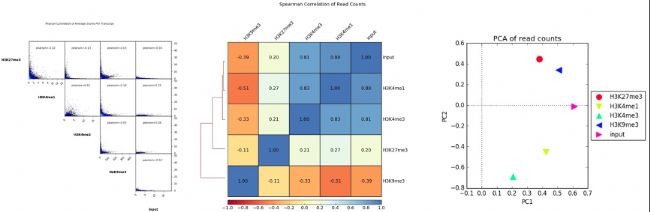 (7)motif分析
(7)motif分析
Motif為一段有特征的DNA短序列,主要為轉錄因子的識別位點,不同的motif對應不同的轉錄因子。
(8)peak的基因注釋和功能分析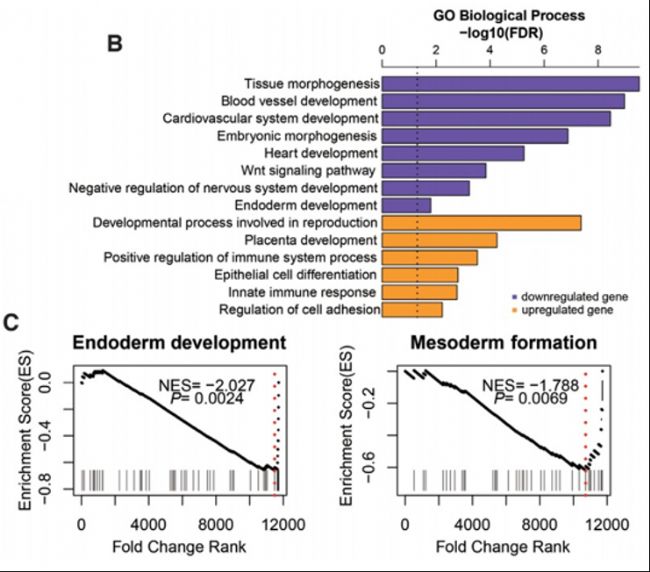
2、差異peak分析
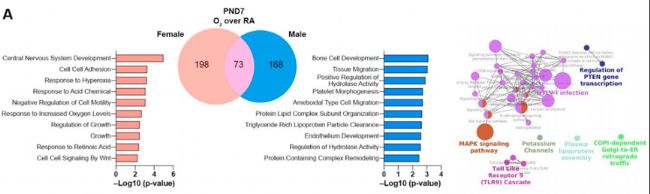
(1)非時間序列數據:
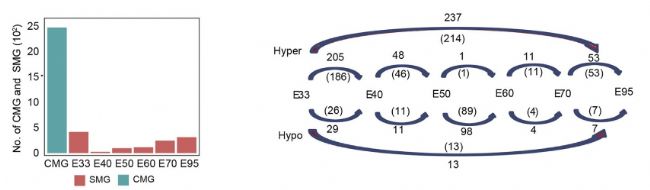
(2)時間序列數據:
(3)差異peak關聯基因的PPI分析
(4)感興趣基因的差異peak展示
3、組學關聯分析:CHIP-seq&轉錄組學
(1)Meta genes整體關聯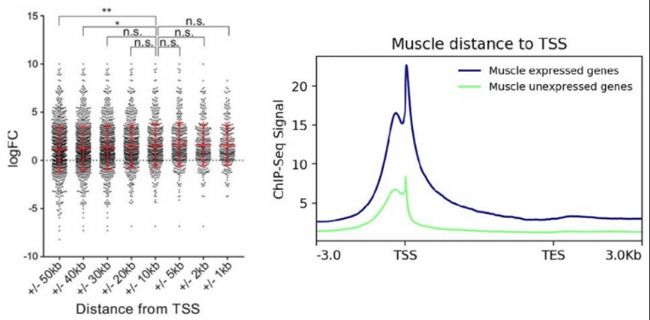
轉錄水平倍數變化 vs. peak倍數變化
(2)差異peak基因-DEG對應關聯:篩選關鍵目的基因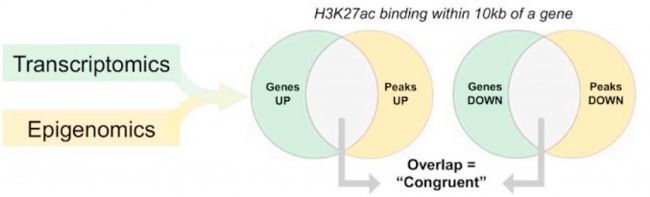
1. 整體把握CHIP-seq圖譜特征:peak/reads在基因組上的分布、peak在元件上的富集、peak在基因元件上的分布、peak的motif分析、peak距離TSS位點的距離分析、peak修飾基因的功能分析
2. 篩選具體差異peak和基因:差異 peak鑒定、非時序數據的分析策略、時序數據的分析策略、差異peak關聯基因的功能分析、差異peak關聯基因的PPI分析、感興趣目標區域的可視化展示
3. CHIP-seq&轉錄組學關聯分析:Meta genes整體關聯、peak關聯基因與DEG對應關聯、目標區域和靶基因的篩選
2. 篩選具體差異peak和基因:差異 peak鑒定、非時序數據的分析策略、時序數據的分析策略、差異peak關聯基因的功能分析、差異peak關聯基因的PPI分析、感興趣目標區域的可視化展示
3. CHIP-seq&轉錄組學關聯分析:Meta genes整體關聯、peak關聯基因與DEG對應關聯、目標區域和靶基因的篩選
后期視情況是否需要下游實驗設計驗證TF結合/組蛋白修飾的目標區域和候選靶基因。
1、圖譜分析
(1)peak/reads在基因組上的分布
l Peak的分布就是蛋白與DNA互作圖譜。
l 不同蛋白對DNA的結合可以按照峰的寬窄和分布特征分為:
· narrow peak:即發生在DNA上特定的短序列,結合的區域很短。
· broad peak:這種類型的peak在DNA上呈彌 散的連續的分布,峰型較寬。
l 一般來說,轉錄因子的峰型都是narrow peak;而對于組蛋白修飾,有的峰型為 narrow peak,有的為broad peak。
· broad peak:這種類型的peak在DNA上呈彌 散的連續的分布,峰型較寬。
· 可以通過調整參數或使用不同的軟件分別鑒定narrow peak及broad peak。
(2)信號的富集程度分析——覆蓋度累積曲線
對樣本比對結果reads累積情況進行展示。一定長度窗口(bin)上reads數進行計數,然后排序,再依次累加畫圖。input (能測到90 DNA片段)在基因組理論上是均勻分布,隨著測序深度增加趨近于直線,實驗組在排序越高的窗口處reads累積速度越快,說明這些區域富集的越特異。
narrow peak :富集程度高;broad peak:富集程度低。
· 富集程度低不代表失敗, 如broad peak。
· 但是如果是轉錄因子, 富集程度低則需要謹慎對待。
· 但是如果是轉錄因子, 富集程度低則需要謹慎對待。
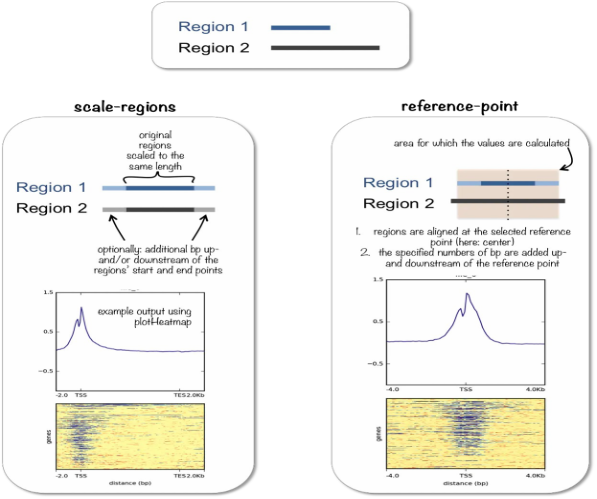
(3)peak/reads的基因元件富集分析
· reference-point(relative to a point): 計算某個點的信號豐度
· scale-regions(over a set of regions): 把所有基因組區段縮放至同樣大小,然后計算其信號豐度。
· scale-regions(over a set of regions): 把所有基因組區段縮放至同樣大小,然后計算其信號豐度。
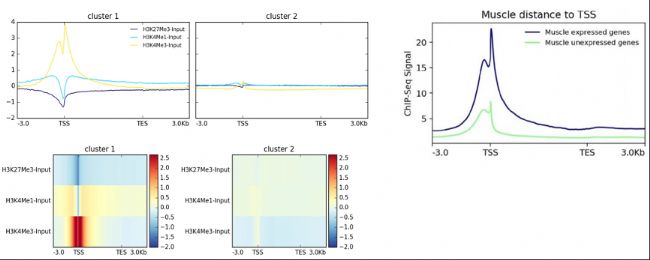
- 基于信號富集的靶基因集分類鑒定(基于聚類算法)
(4)peak/reads的基因元件分布分析
(5)peak/reads與TSS的相對距離分布
轉錄因子、組蛋白修飾往往具有重要的轉錄調控功能,而TSS附近是主要的轉錄調控區域,因此判斷peak與TSS的位置關系有重要的意義。
(6)降維分析
將基因組分為等長窗口(bins),計算各樣本各窗口內的Reads覆蓋情況并進行標準化。基于此數據進行相關性、聚類和PCA分析。
Motif為一段有特征的DNA短序列,主要為轉錄因子的識別位點,不同的motif對應不同的轉錄因子。
· 根據motif可以推測結合的轉錄因子。
· 已知轉錄因子則分析該轉錄因子識別的序列特征。
· 已知轉錄因子則分析該轉錄因子識別的序列特征。
(8)peak的基因注釋和功能分析
· ORA
· GSEA: 可以按照peak信號強度排序
· GSEA: 可以按照peak信號強度排序
2、差異peak分析
(1)非時間序列數據:
(2)時間序列數據:
(3)差異peak關聯基因的PPI分析
(4)感興趣基因的差異peak展示
3、組學關聯分析:CHIP-seq&轉錄組學
(1)Meta genes整體關聯
- 距離TSS位點不同距離的peak注釋到的基因的表達水平分析
- 不同表達水平的基因,peak的數量分布對比
轉錄水平倍數變化 vs. peak倍數變化
(2)差異peak基因-DEG對應關聯:篩選關鍵目的基因
- peak關聯基因與差異表達基因的重疊分析。
- peak關聯基因可以是peak注釋到啟動子區,TSS±10kb區的基因,也可以來自已 知公共數據庫的注釋,如Human Enhancer Disease Database (HEDD)。
- 九象限圖法
標簽:
ChIP-seq




Copyright(C) 1998-2025 生物器材網 電話:021-64166852;13621656896 E-mail:info@bio-equip.com