
GSK:使用單一DNA結構快速構建穩定的慢病毒載體生產細胞系
慢病毒載體(Lentiviral vectors, LVs)是以人類免疫缺陷型病毒(HIV)為基礎發展起來的基因治療載體,最被廣泛應用的慢病毒載體是基于雙鏈RNA病毒人類免疫缺陷I型病毒(HIV-1)[1]。HIV-1型慢病毒載體系統的建立,經歷了一個逐步完善的過程,其生物安全性不斷提高。
LVs對分裂細胞和非分裂細胞均具有感染能力,更加安全,并可以在體內長期的表達。這一特性使LVs成為臨床研究的理想選擇,美國食品和藥物管理局(FDA)和歐洲藥物管理局(EMA)已經批準多個以慢病毒為基礎的基因療法。目前,LVs在β-地中海貧血(β-thalassemia)、鐮狀細胞性貧血(Sickle cell anemia)、實體瘤(Solid tumors)和COVID-19等疾病(圖1)的細胞和基因治療中得到了廣泛的應用,如CAR-T細胞治療。
臨床試驗需要大量功能性慢病毒顆粒,提高慢病毒的包裝與生產水平可有效促進慢病毒介導的基因治療的發展。LVs通常是通過將編碼載體組分的多質粒瞬時轉染到貼壁HEK293T細胞中來生產。瞬時轉染的方式需要耗費很高的成本和很長的時間來獲取質粒DNA,從而使生產過程極其昂貴;在放大過程中,質粒和轉染試劑的殘留,也會引起終產物的污染。相較于瞬時轉染生產,穩定的慢病毒生產才是基因治療的更優選擇。穩定生產細胞株將其所有載體編碼的DNA都穩定地整合到宿主細胞基因組中,可以降低生產成本、提高整體安全性和可重復性。
英國著名的葛蘭素史克藥物研究中心報告了一種通過穩定轉染編碼所有慢病毒載體成分的單個 DNA 結構體到 293T 細胞,快速生成穩定生產慢病毒載體的細胞系的方法。由此產生的穩定懸浮細胞系可生產出與瞬時轉染一樣高滴度的慢病毒,可以在一次性攪拌式生物反應器中輕松放大,并且在后續細胞培養中遺傳和功能穩定。通過在慢病毒載體的上游加工過程中消除對有效瞬時轉染的要求,并改用固有的可擴展懸浮細胞培養形式,他們認為這種方法將生產出比當前制造工藝更高的批量產量,并使患者能夠更好地獲得基于慢病毒載體的藥物[3]。
載體構建
LVs生產細胞株通常是基于順序穩定轉染或將編碼每個載體組分(轉移質粒、包裝質粒gagpol-rev和包膜質粒VSVg)的DNA轉導到基因組不同位點的宿主細胞。這種策略導致細胞系開發時間長,并且至少有一個基因座可能發生遺傳或轉錄不穩定的高風險,從而導致生產力損失。為了避免這種情況,GSK使用第三代慢病毒載體系統,載體組件由四個獨立的轉錄單元編碼,每個單元都有自己的啟動子和多聚腺苷酸化信號,防止產生復制能力強的慢病毒(RCL)顆粒,一個將所有的載體成分構建成單個大DNA結構(BAC)并整合入宿主細胞(圖2)。
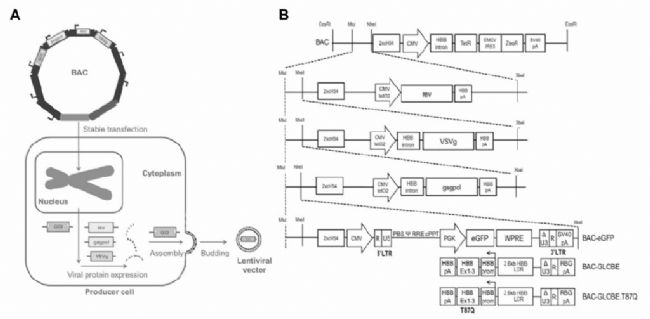
慢病毒載體穩定生產細胞系的開發
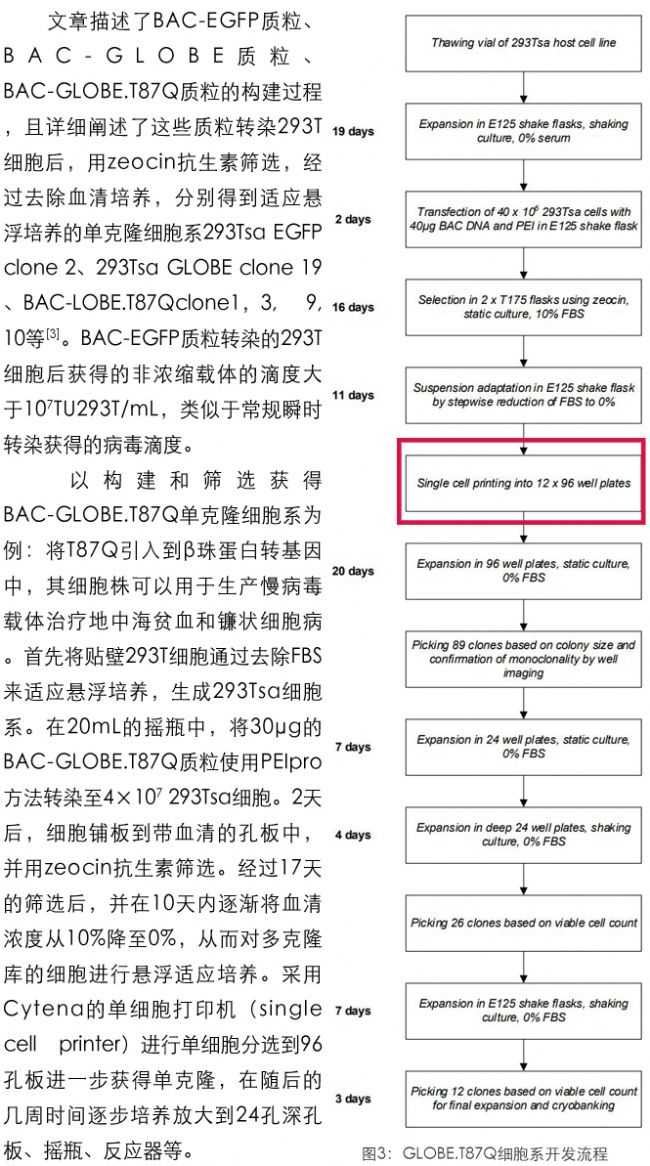
慢病毒載體的穩定生產
利用微滴式數字PCR (ddPCR)定量每個載體成分DNA拷貝數,每個成分的拷貝數在1到10拷貝數/細胞之間變化(圖4A)。通過靶向位點擴增技術(Targeted locus amplification,TLA)對293Tsa EGFP clone 2的BAC DNA進行整合位點定位,在chr3:169,967,149-169,967,179處發現了一個整合位點(圖4B),在293Tsa GLOBE clone 19中,于chrl: 146,557,007和chrX: 127,363,436處發現了兩個整合位點(圖4C),表明BAC DNA在隨機整合到宿主細胞基因組之前發生了異位串聯,轉染圓形的BAC DNA質粒,宿主的DNA修復蛋白會對其進行重排,從而產生線性整合DNA。
以MOI=50或100的慢病毒轉導CD34+細胞,通過qPCR檢測轉導細胞的載體拷貝數(VCN),重復轉導使得每個細胞產生2個拷貝的VCN,表明穩定的細胞系產生的病毒載體可以感染類似難以感染的原代細胞(圖4D)。
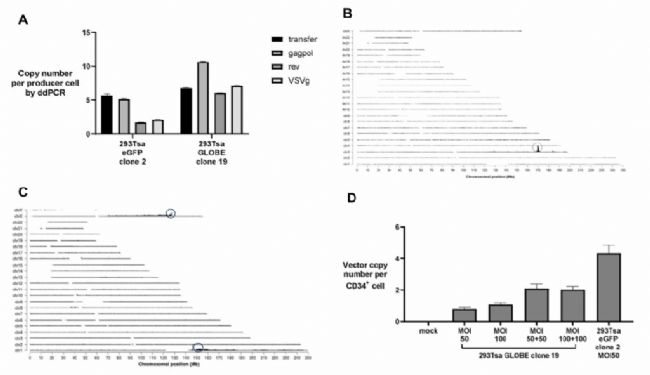
慢病毒穩定包裝和瞬時轉染生產對比
構建相同的4質粒系統瞬時轉染293Tsa細胞,通過RNA測序(RNA-seq)來定量各載體組分的RNA水平。各組分的RNA瞬時表達和穩定表達之間沒有顯著差異,只有293Tsa GLOBE clone 19的rev表達升高(rev比瞬時表達高2.88倍,p = 0.0003);并且,穩定克隆的不同培養瓶之間每個載體組分的RNA表達水平的變異性顯著低于瞬時表達(p < 0.001) (圖5A)。另外,瞬時轉染和穩定表達的慢病毒在病毒滴度效價方面均未觀察到顯著差異(圖5B)。

穩定包裝生產慢病毒的功能和遺傳穩定性驗證
使用低溫儲存的12個單克隆細胞進行排序,在15ml一次性攪拌槽微生物反應器中制備慢病毒載體并檢測其病毒滴度(圖6A)。選取效價最高的4個克隆(clone1, 3, 9, 10)連續搖瓶培養數周來評估病毒滴度和遺傳穩定性,使用選取三個時間點,在強力霉素誘導下,使用ddPCR和ELISA法對比病毒滴度,clone3, 9, 10的病毒滴度沒有顯著變化,clone1的滴度下降(p = 0.0011)(圖6B);在相同的三個時間點,采集非誘導的細胞基因組DNA檢測BAC各基因拷貝數量變化,四個克隆在遺傳上都是穩定的,既沒有丟失整合DNA編碼的載體成分,也沒有通過重復感染擴增轉移載體拷貝數(VCN)(圖6B)。

具有復制能力的慢病毒(RCL)的檢測
為了測定這些克隆細胞是否會產生RCL,采用產物增強逆轉錄酶(PERT)檢測了12個樣品,這12個樣品分別來自293Tsa EGFP clone 2、GLOBE clone 19和GLOBE.T87Q clone 1、3、9和10這6個克隆細胞的的收集樣品和濃縮后樣品。結果顯示,這些穩定生產的細胞系中都沒有檢測到RCL(表1)。

總結
在GSK分享的單DNA分子BAC結構中將CMV啟動子驅動的轉錄單元以transfer-gagpol-VSVg-rev的順序首尾1:1:1:1相連排列,但是也發現,在相同的BAC轉染細胞后,不同的單克隆細胞的每個載體基因DNA拷貝數和mRNA表達水平存在差異。因此,在細胞系構建的過程中,應篩選足夠數量的克隆,以獲得一個mRNA表達水平理想的克隆,獲得最佳的產量和感染性。將BAC結構轉染宿主細胞后,根據經驗能夠從幾十個單克隆細胞系中篩選出高產的克隆,因此GSK認為,這種包裝細胞系的方法能夠減少克隆之間變異。未來也可以進一步的載體改造,將其中的基因片段用改進的基因進行替換,快速利用多個片段組裝或基因合成為一個BAC片段,單次的轉染能簡化并加速慢病毒穩定生產細胞株的構建流程,提高慢病毒載體的產量、質量和安全性。FDA和EMA也認為這種穩定生產慢病毒的細胞系平臺可用于生產臨床使用的慢病毒,也沒有提出慢病毒載體制造工藝之外的要求。這種簡單有效的生產細胞株培養技術,可廣泛應用于慢病毒載體制造的產業化,并有可能在未來為更多的患者提供基因治療藥物。
瞬時轉染為基礎的慢病毒載體制造工藝用于臨床是基因治療領域的標準方法,但通常需要大量GMP級別的質粒DNA和轉染試劑。相比較之下,構建穩定的生產細胞株更適合于大規模的慢病毒的生產,建高產細胞株的主要流程如圖6所示,經過單克隆細胞篩選、擴增培養和驗證,最終獲得細胞活力高、生長能力強,并且表達水平、穩定性、質量情況都具有最高表現的單克隆細胞株。

GSK利用Cytena單細胞打印機替代常規的有限稀釋的方法篩選單克隆細胞,由于其溫和而高效的分選原理,使得單細胞率>95%,克隆恢復率與有限稀釋相當,獲得大量單克隆來源的細胞,并提供分選過程中的噴嘴圖片作為單克隆細胞來源的佐證,進一步加速慢病毒生產細胞株構建的工作流程,促進基因與細胞治療的發展。

參考文獻
[1].Meng F, Chen C, Wan H, Zhou Q. [Advances of lentiviral vectors]. Zhongguo Fei Ai Za Zhi. 2014 Dec;17(12):870-6. Chinese. doi: 10.3779/j.issn.1009-3419.2014.12.09. PMID: 25539614; PMCID: PMC6000409.
[2].Ferreira MV, Cabral ET, Coroadinha AS. Progress and Perspectives in the Development of Lentiviral Vector Producer Cells. Biotechnol J. 2021 Jan;16(1):e2000017. doi: 10.1002/biot.202000017. Epub 2020 Aug 2. PMID: 32686901.
[3].Chen YH, Pallant C, Sampson CJ, Boiti A, Johnson S, Brazauskas P, Hardwicke P, Marongiu M, Marinova VM, Carmo M, Sweeney NP, Richard A, Shillings A, Archibald P, Puschmann E, Mouzon B, Grose D, Mendez-Tavio M, Chen MX, Warr SRC, Senussi T, Carter PS, Baker S, Jung C, Brugman MH, Howe SJ, Vink CA. Rapid Lentiviral Vector Producer Cell Line Generation Using a Single DNA Construct. Mol Ther Methods Clin Dev. 2020 Aug 14;19:47-57. doi: 10.1016/j.omtm.2020.08.011. PMID: 32995359; PMCID: PMC7501408.



