
低流體剪切應力激活的Smad2/3信號通路介導動脈向內重塑
脊椎動物血管系統的設計使局部灌注與每個組織的代謝需求相匹配,從而優化功能并最終存活。例如,缺氧會在短時間內誘導擴張小阻力血管因子的分泌,而在長時間內則會誘導刺激血管生成因子的分泌。這些調整增加了受影響組織的大動脈的流量,誘導其向外重塑。相反,組織的萎縮會導致血管密度降低和供血動脈向內重塑。
排列在血管內表面的內皮細胞(ECs)傳遞來自血流動力學壁流體剪切應力(FSS)的信號以調節血管重塑。多項研究表明,增加或減少動脈血流的手術干預分別刺激向外或向內重塑。目前的證據支持內皮細胞編碼FSS設定值介導體內平衡的模型。持續高于或低于這個值的FSS會引起向外或向內的重塑,從而使FSS恢復到原始值。相比之下,設定值附近的FSS穩定了血管。不同類型的內皮細胞的設定值不同,對應于特定血管類型的剪切應力生理水平。對于人臍靜脈ECs(HUVECs),生理 FSS 在 10-20 dynes/cm2 的范圍內,抑制炎癥性NF-κB,而Smad1/5信號在此范圍內被最大程度激活。
轉化生長因子(TGF)-β/骨形態發生蛋白(BMP)家族成員通過信號傳導調控細胞反應,包括從早期發育階段到成年生活和疾病的增殖,分化和遷移。已經表明,TGF-β/BMP家族配體和受體在剪切應力調控血管穩態中的作用。
耶魯大學心血管研究中心、羅格斯大學高級生物技術和醫學中心、上海市免疫學研究所等研究團隊以前的研究表明,Smad1/5在生理設定值附近的激活和核易位是由其受體Alk1和內皮糖蛋白觸發的。流動激活是通過對循環TGF-β家族配體BMP9和BMP10的敏感性增加約20倍介導的。與血管穩定的作用一致,這些受體的突變或缺失會誘發脆弱且容易破裂的血管畸形。其他研究表明,相關的Smad2/3通路也被血流激活,但是,在這種情況下,激活被生理層流FSS抑制,并由振蕩FSS激活,這是動脈粥樣硬化易感區域的典型特征。這些結果促使該團隊研究了Smad2/3信號在血流依賴性血管重塑中的作用。

剪切應力激活Smad2/3
用1-30 dynes/cm2 的FSS 處理 HUVECs 持續12 小時誘導 Smad2/3 核易位,其在低 FSS(1-5 dynes/cm2)下達到最大值,然后隨著FSS達到生理范圍(≥12 dynes/cm2)后顯著降低(圖1 A、B)。細胞排列的平行評估(圖1 C)證實了對FSS的預期大小反應。兩個Smad2/3 直接靶基因—纖連蛋白和整合素α5 的 qPCR 表明,它們的表達遵循相似的大小依賴性,在低 FSS 時達到最大值(圖1 D、E)。這種反應被小干擾RNA(siRNA)介導的Smad2/3 缺失阻斷,證實了它們對Smad2/3活性的依賴性。因此,ECs中的Smad2/3信號顯示雙相調節,在低FSS時最大。
TGF-β受體通常通過關鍵絲氨酸殘基的C端磷酸化啟動R-Smad信號傳導。因此,實驗研究了剪切應力強度(1-30 dynes/cm2)在12小時內對 Smad2/3 C 末端磷酸化(p-Smad2/3 C)的影響。在 12 小時的時間過程中,p-Smad2/3 在流動開始后約 2-4 小時達到峰值,然后下降到高于基線的平臺期(圖1 F)。然而,與核易位不同,p-Smad2/3在高FSS下沒有降低。因此,雖然流動誘導p-Smad2/3 C,但它不像核易位那樣對FSS大小表現出相同的雙相依賴性。因此,在高FSS下抑制核定位和基因誘導必須涉及一種獨特的機制。
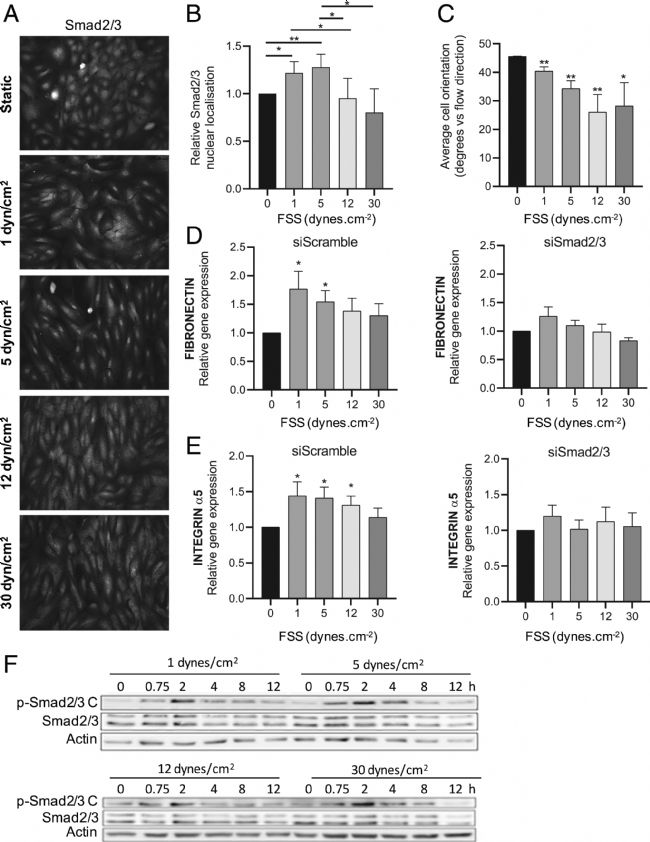
圖1 Smad2/3受剪切應力調節。
受體和配體的鑒定
為了確定哪些受體介導Smad2/3的流動激活,首先考慮了Alk5(TGFβR1),這是TGF-β的主要I型受體,還考慮了神經纖毛蛋白-1(Nrp1),基于最近的一份報告,它與Alk1和Alk5相互作用,并參與VEGFR2的FSS激活。結果表明,Alk5 和 Nrp1 是 Smad2/3 流動激活所需的關鍵受體,就像 Alk1 和內皮糖蛋白是 Smad1/5 流動激活所需的關鍵受體一樣。
通過類比Smad1/5的流動激活中對BMP9或BMP10的要求,接下來討論了可溶性配體的作用。無血清培養基中FSS未能激活細胞中Smad2/3,表示需要一個或多個循環因子。TGF-β是刺激Smad2/3活化的主要Alk5配體。然而,濃度為1 pg/mL-10 ng/mL的TGF-β2與剪切應力沒有協同作用。由于流動增強了BMP9或BMP10對Smad1/5的激活,于是接下來測試了這些配體。結果表明,FSS對Smad2/3活化的增強是由于對BMP9而非相關配體的敏感性增加。
誘導的體內向內重塑
接下來使用部分頸動脈結扎模型檢查了體內低流量介導的向內重塑。兩個頸動脈分支的結扎(圖2 A)急劇減少了血流,這通常導致內膜-內側增厚和管腔面積減少30%至40%(圖2 B)。對側頸動脈血流也有代償性增加,但向外重塑要少得多。實驗首先討論了低FSS是否在體內激活了Smad2/3。大鼠和小鼠的頸動脈結扎(圖2 C、D)后頸動脈上游ECs中的Smad2/3核染色增加。通過EC特異性敲除(ECKO)Alk5降低了小鼠的這種作用。因此,Smad2/3在體內被低FSS激活,這需要Alk5。
此外,阻斷Smad2/3激活是否會減少低流動誘導的動脈向內重塑。在野生型小鼠中,右頸動脈周長減少了18%,對應面積減少了33%,而Alk5 ECKO小鼠沒有顯著變化(圖2 E)。這些數據表明,Alk5-Smad2/3信號通路是低流動誘導的向內動脈重塑所必需的。
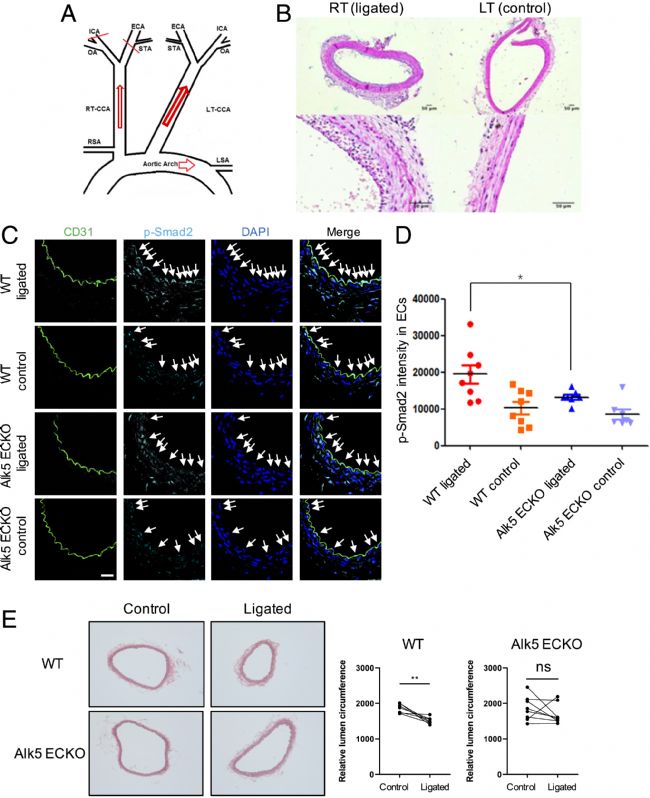
圖2 低流動誘導的體內向內重塑。
高FSS下的抑制
Smad2/3 的雙相流動激活表明,在高 FSS 下激活的另一種通路拮抗 p-Smad2/3 核易位。敲除小鼠MEKK3通過激活TGFβR1-Smad2/3通路觸發自發的向內動脈重塑。MEKK2 和 MEKK3 是在高 FSS 下誘導轉錄因子 Klf2 表達的途徑中的上游組分。該通路介導eNOS和其他促進血管舒張和穩定血管系統的基因表達。因此,研究了siRNA介導的MEKK3缺失對Smad2/3信號流動激活的影響。敲低MEKK3在低剪切下影響不大,但在高FSS下消除了Smad2/3核轉運的抑制(圖3 A、B)。因此,高-流量抑制Smad2/3功能需要MEKK3。
接下來,測試了Klf2在這條通路中的作用。敲低Klf2后,ECs受到不同幅度的FSS。同樣,Klf2缺失在低FSS下幾乎沒有影響,但在高FSS下阻止Smad2/3的抑制(圖3 C、D)。eNOS是一種經過充分驗證的Klf2靶基因。兩種不同的抗eNOS siRNAs在高流量條件下對eNOS幾乎完全敲低,但對Smad2/3核定位沒有影響。這一結果表明,必須有其他靶基因介導Smad2/3活性的抑制作用。然而,Klf2調控了數百個EC基因的表達,這使得鑒定具有挑戰性。因此需要更仔細地研究高剪切下核排斥機制。

圖3 MEKK3和Klf2在高FSS下抑制Smad2/3。
Smad2/3連接區磷酸化
Smad2/3抑制的一個重要機制涉及連接N端MH1和C端MH2結構域的“連接區”區域中絲氨酸和蘇氨酸殘基的磷酸化。因此,實驗測量了低FSS和高FSS下的Smad2/3連接區 的磷酸化。結果表明,在高FSS下,多個連接區域絲氨酸的磷酸化顯著增加。為了確定這種機制是否介導高FSS下的核排斥,轉染了四個關鍵殘基中攜帶Smad3突變的細胞。該突變體在高剪切下保存細胞核。接下來又測試了MEKK3和Klf2在連接區磷酸化中的作用。任何一種蛋白質的缺失都減少了高FSS誘導的連接區磷酸化。這些數據共同表明,通過MEKK3-Klf2通路的高流量誘導的Smad2/3連接區磷酸化介導了高剪切下Smad2/3核易位的抑制。
已知的 Smad 連接區激酶包括絲裂原活化蛋白激酶(Erk、Jnk 和 p38)、周期蛋白依賴性激酶(CDKs)、Rho相關蛋白激酶Akt、鈣調素依賴蛋白激酶和糖原合酶激酶-3。然而,實驗注意到CDK被Klf2抑制,并預測靶向CDKs將在高剪切下被激活,從而可能介導觀察到的Smad連接區磷酸化。最近的研究表明,高FSS下的動脈ECs處于G1停滯晚期,表明CDK4或CDK2的激活。因此,實驗專注于CDK2和CDK4。CDK2的敲低在高剪切下強烈阻斷細胞中的核排斥,而CDK4 敲低幾乎沒有影響。此外,CDK2 敲低阻斷連接區磷酸化。綜合這些結果,得出結論,CDK2是連接區磷酸化所必需的。
接下來評估了Klf2調控的CDK抑制劑(CDKN2B、CDKN1A)的作用。CDKN2B的敲低而不是CDKN1A的敲低逆轉了高流量下ECs中Klf2缺失的影響。因此,MEKK3-Klf2通路的CDKN2B下調,隨后CDK2激活,介導Smad2/3連接區磷酸化和高FSS抑制。
CDK在體內的抑制作用
最后,研究了該通路是否在體內起作用。黃酮吡醇已被用于癌癥患者的人體臨床試驗。然而,這種化合物在循環中的半衰期很短(∼30分鐘)。注射高劑量(7.5 mg/kg)黃酮吡啶醇的小鼠肺動脈和股動脈的內皮顯示p-Smad2核染色增加數倍(圖4 A-D)。這些結果證實了CDKs在動脈血流下阻止Smad2/3核易位中的作用。接下來,討論了這種處理是否會引起血管重塑。結果發現,注射3天中的2天的小鼠存活率更高,因此,根據該方案注射3周。肺血管系統在MEKK3 敲低后最容易重塑。對整個肺切片的檢查顯示,血管的平滑肌肌動蛋白(SMA)覆蓋率顯著增加(圖4 E),右心室血壓顯著升高(圖4 F)。
這些數據共同定義了一條通路,其中MEKK3-Klf2-CDK2通過Smad2/3連接區磷酸化抑制高FSS下的Smad2/3核易位,以限制生理流大小的向內重塑。
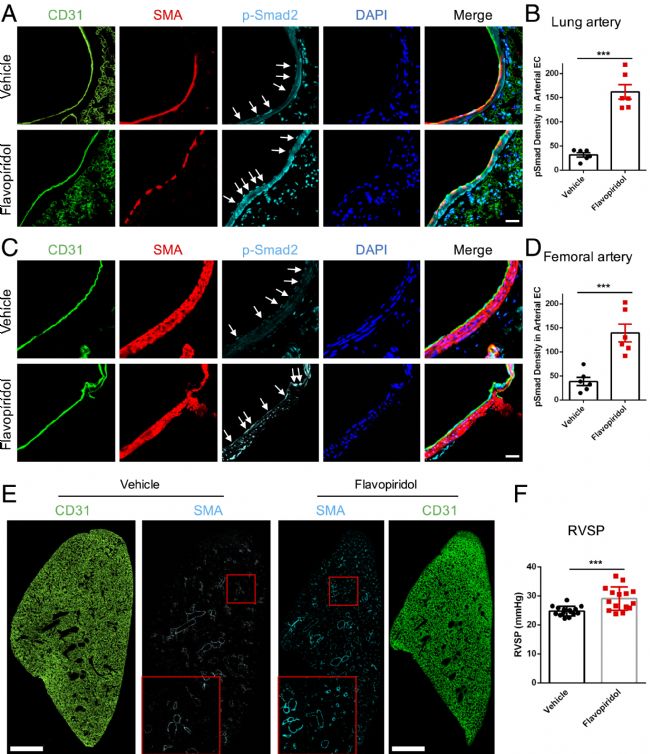
圖4 CDK在體內的抑制作用。
總之,該文分析了調節動脈管腔大小的信號網絡,從而分析了它們將血液輸送到組織的能力。管腔直徑處于嚴密的穩態調節下,以使動脈血流與下游組織的需求相匹配。然而,在動脈粥樣硬化中,這種調控受到破壞,導致血流受限和組織缺血。闡明Smad2/3通路在低流量下被特異性激活以觸發向內重塑,從而恢復正常的剪切應力水平,不僅揭示了生理調節的機制,而且還解釋了病變動脈中炎癥因子激活同一通路如何導致動脈受限。
參考文獻:Deng H, Min E, Baeyens N, Coon BG, Hu R, Zhuang ZW, Chen M, Huang B, Afolabi T, Zarkada G, Acheampong A, McEntee K, Eichmann A, Liu F, Su B, Simons M, Schwartz MA. Activation of Smad2/3 signaling by low fluid shear stress mediates artery inward remodeling. Proc Natl Acad Sci U S A. 2021 Sep 14;118(37):e2105339118. doi: 10.1073/pnas.2105339118. PMID: 34504019; PMCID: PMC8449390.
原文鏈接:https://pubmed.ncbi.nlm.nih.gov/34504019/
小編旨在分享、學習、交流生物科學等域的研究進展。如有侵權或引文不當請聯系小編修正。
微信搜索公眾號“Naturethink”,了解更多細胞體外仿生培養技術及應用。
產品:
小型流體剪切力細胞培養儀
http://www.xhtechnology.cn/show1equip4841798.html
動脈壁面剪切力多細胞共培養裝置
http://www.xhtechnology.cn/show1equip4891724.html
上皮與間充質轉化流體剪切力裝置



