
小鼠模型在揭示MTHFD2通過抑制PTEN調控巨噬細胞極化研究中的應用
巨噬細胞在體內發揮著不同的功能,以響應各種微環境刺激。近年來研究發現,巨噬細胞可根據微環境的變化而改變其表型,這被稱為巨噬細胞極化。M1型巨噬細胞通常由IFN-γ或LPS或TNF-α激活,具有細胞毒性、促炎和抗腫瘤功能。M2型巨噬細胞則由IL-4或IL-13或IL-10交替激活,發揮清除寄生蟲、抗炎和促腫瘤作用。
巨噬細胞的極化受轉錄因子、信號級聯和代謝重編程的調控。一碳(1C)代謝包括葉酸循環和蛋氨酸循環等,在多種生理過程中發揮重要作用。一碳代謝酶MTHFD2(亞甲基四氫葉酸脫氫酶2)參與了腫瘤發生和免疫細胞功能的調控,但它是否能調控巨噬細胞極化目前仍是一個未知數。
近日,天津醫科大學基礎醫學院免疫系余秋景教授(現為電子科技大學附屬醫院·四川省人民醫院健康管理中心教授)和藥理系王霆教授以及天津醫科大學第二醫院陳兵教授領導的團隊通過研究發現,MTHFD2是巨噬細胞極化的調節因子。它通過與PTEN催化中心相互作用調節PTEN活性,從而對巨噬細胞極化進行重編程。這項成果發表在《Cell Reports》雜志上,表明操控MTHFD2可作為一種策略來調節巨噬細胞介導的免疫反應。

圖片來源:《Cell Reports》
(https://doi.org/10.1016/j.celrep.2023.112481)
研究材料
在此次研究中,研究人員將賽業生物提供的Mthfd2fl/fl小鼠與Lyz2-Cre小鼠雜交,培育出髓系細胞特異性MTHFD2敲除小鼠(MTHFD2-KO-Mφ小鼠)。在此基礎上,通過構建CCl4誘導的肝纖維化模型、甲殼素模型和腫瘤移植模型。他們從小鼠身上采集骨髓,并使其分化為骨髓來源的巨噬細胞(BMDM)。他們通過免疫沉淀分析檢測到MTHFD2與PTEN的相互作用,并通過ClusPro蛋白對接分析、鏈霉親和素pull down和表面等離子體共振(surface plasmon resonance, SPR)等實驗確定了相互作用結構域和關鍵位點。
技術路線
01 通過體外實驗探究MTHFD2如何調控小鼠巨噬細胞的極化
02 通過多個模型確定MTHFD2在體內可抑制M1型巨噬細胞極化,并促進M2型極化
03 通過對Akt信號轉導的分析發現MTHFD2可抑制PTEN的PIP3磷酸酶活性
04 蛋白相互作用分析表明,MTHFD2(aa 215-225)直接靶向PTEN催化中心(aa 118-141),且D168殘基對這種相互作用很重要
研究結果
1.MTHFD2在體外和體內調節巨噬細胞的極化
研究人員首先分析了MTHFD2的表達和功能是否與巨噬細胞極化有關。在M1和M2極化模型中,MTHFD2的表達均升高,表明它可能是巨噬細胞極化的重要調節因子。利用siRNA抑制小鼠巨噬細胞系RAW264.7中的MTHFD2后,M1型標志物Nos2和Il-6表達顯著上調,而M2型標志物Arg1和Retnla表達顯著下調。
研究人員將賽業生物提供的Mthfd2fl/fl小鼠與Lyz2-Cre小鼠雜交,培育出髓系細胞特異性MTHFD2敲除小鼠(MTHFD2-KO-Mφ小鼠)。他們發現,與WT小鼠(MTHFD2-WT-Mφ小鼠)相比,KO小鼠(MTHFD2-KO-Mφ小鼠)BMDMs中的M1型標志物上調,而M2型標志物被抑制。MTHFD2過表達則抑制M1型標志物,但增強M2型標志物表達。這些結果表明,MTHFD2可以調節巨噬細胞的極化。
為了進一步研究髓系MTHFD2是否調節體內巨噬細胞的極化,研究人員在MTHFD2-KO-Mφ和MTHFD2-WT-Mφ小鼠上建立了腫瘤移植模型。與WT小鼠相比,KO小鼠的腫瘤生長較慢,腫瘤切片中的F4/80+巨噬細胞數量增加了近2倍(圖1)。表達iNOS的細胞數量明顯增加,而表達ARG1的細胞數量減少,這表明Mthfd2缺失會促進M1型巨噬細胞極化,并抑制M2型巨噬細胞極化。
由于表達ARG1的巨噬細胞在促進纖維化緩解方面發揮重要作用,故研究人員利用CCl4誘導的肝纖維化模型來分析MTHFD2在調節M2表型上的作用。MTHFD2缺失導致ARG1陽性的巨噬細胞顯著減少,表明MTHFD2對M2表型有正向調節作用。此外,通過甲殼素模型的分析,他們發現髓系MTHFD2缺失會減少嗜酸性粒細胞的招募并降低ARG1的表達,進一步提示了MTHFD2在促進M2型巨噬細胞功能上發揮重要作用。

圖1 MTHFD2在體內調節腫瘤相關巨噬細胞的極化[1]
2.MTHFD2抑制PTEN的PIP3磷酸酶活性
之前的研究發現,Akt-mTORC1信號傳導對IL-4誘導的M2型極化至關重要。有意思的是,在MTHFD2-KO BMDM中,AktS473和AktT308的基礎磷酸化及IL-4誘導的磷酸化都明顯減弱。那么,Akt激活的減少是否與M2型極化缺陷有關?研究人員用Akt激活劑SC79處理MTHFD2-KO BMDM后,發現巨噬細胞極化的缺陷得以挽救,表明Akt信號轉導對MTHFD2缺失誘導的異常極化很關鍵。
蛋白激酶PI3K和PTEN可通過平衡PIP3的濃度來調節Akt的激活。他們發現,在經過IL-4和IFN-γ處理的MTHFD2-KO和WT細胞中,PI3K的表達和活性相當,PTEN表達也相當。然而在基礎條件和IL-4處理條件下,MTHFD2-KO細胞中PTEN的PIP3磷酸酶活性均高于WT細胞。與此相一致的是,PTEN的敲除可明顯挽救MTHFD2-KO細胞中的Akt磷酸化。綜上,這些結果表明,MTHFD2抑制了PTEN的PIP3磷酸酶活性,從而促進了Akt的激活。
3.MTHFD2直接靶向PTEN催化中心
通過共聚焦免疫熒光顯微鏡觀察和免疫沉淀分析,研究人員發現MTHFD2能夠與PTEN發生相互作用。通過構建MTHFD2的N端缺失突變體(缺少線粒體靶向信號),他們發現MTHFD2與PTEN的結合以及PTEN的PIP3磷酸酶活性和Akt信號轉導均不受影響。此外,IL-4刺激明顯增強了這種相互作用,而IFN-γ處理則沒有。由此可見,線粒體定位并不是MTHFD2與PTEN結合的必要條件,且IL-4處理促進了這種相互作用。
通過ClusPro蛋白對接分析,他們進一步發現MTHFD2的氨基酸殘基(aa 215-225)可直接靶向PTEN的催化中心(aa 118-141)(圖2)。鏈霉親和素pull-down和SPR分析發現,合成的PTEN(aa 118-141)生物素肽段能夠與胞內或重組MTHFD2蛋白相互作用。同樣地,MTHFD2(aa 215-225)生物素肽段也能與胞內或重組PTEN蛋白相互作用。與WT MTHFD2相比,缺乏aa 215-225的MTHFD2能夠顯著減少PTEN的PIP3磷酸酶活性降低。這些結果表明,MTHFD2直接靶向PTEN催化中心以降低PTEN的PIP3磷酸酶活性。
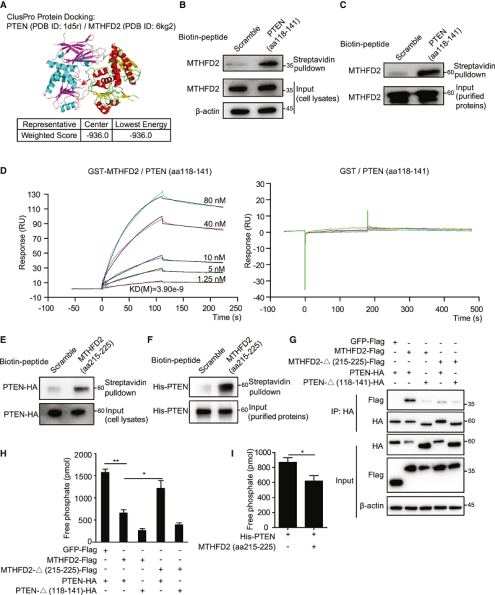
MTHFD2直接靶向PTEN催化中心并抑制PTEN的PIP3磷酸酶活性[1]
MTHFD2需要無機磷酸鹽和鎂離子來支持其依賴于NAD的脫氫酶活性,而以往的研究發現MTHFD2 D168是鎂離子結合位點,而R201是無機磷酸鹽與NAD結合的主要位點。因此,研究人員通過突變分析來確定這些結合位點對MTHFD2與PTEN相互作用的重要性。他們發現,D168E突變體與PTEN的結合明顯減少。在MTHFD2-KO BMDM中,過表達MTHFD2可使M1型和M2型標志物恢復到WT細胞的水平,但過表達D168E突變體只能部分恢復表型。從這些結果可以看出,MTHFD2 D168對于MTHFD2與PTEN的結合并抑制PTEN的PIP3磷酸酶活性很關鍵。
研究結論
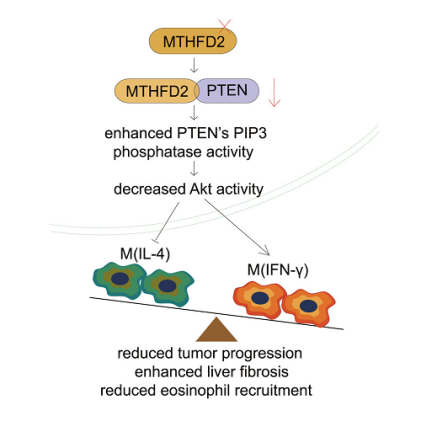
圖3 MTHFD2調節巨噬細胞極化的機理示意圖[1]
總的來說,這項研究通過體外和體內研究揭示了MTHFD2的一個重要非代謝功能:它通過與PTEN的催化中心相互作用來調節PTEN活性,從而重編程巨噬細胞極化并改變巨噬細胞介導的免疫反應(圖3)。它表明調控MTHFD2可作為一種策略來調節巨噬細胞介導的免疫反應。
原文檢索:
[1]Shang, M., Ni, L., Shan, X., et al. MTHFD2 reprograms macrophage polarization by inhibiting PTEN. Cell Rep. 2023 May 30;42(5):112481. https://doi.org/10.1016/j.celrep.2023.112481



