
mRNA-seq在研究炎性巨噬細胞活化和類風濕關節炎治療方法中的應用

期刊:MedComm (2020)
影響因子:9.9
伯豪產品服務:mRNA-seq
導讀
在類風濕關節炎(RA)的發病機制中,炎癥巨噬細胞的表觀遺傳調控控制炎癥的發生和消退。然而,巨噬細胞介導的關節炎損傷的機制在很大程度上仍然不清楚。本研究發現,在RA患者和實驗性關節炎小鼠中,滑膜組織中賴氨酸乙酰轉移酶2A (KAT2A)的表達增加與炎癥性關節免疫病理密切相關。MB-3 (KAT2A特異性化學抑制劑)可顯著改善膠原誘導關節炎模型中的滑膜炎和骨破壞。藥理抑制和siRNA沉默KAT2A不僅抑制了先天刺激觸發的促炎基因(如Il1b和Nlrp3)的轉錄,而且在體內和體外也破壞了Nlrp3炎性體的激活。機制上,KAT2A通過抑制NRF2活性及下游抗氧化分子促進巨噬細胞糖酵解重編程,支持H3K9ac,限制NRF2介導的促炎基因轉錄抑制。靶向KAT2A代表了RA和相關炎性疾病患者的潛在治療方法。
研究技術
mRNA-seq
技術路線圖
研究結果
1. 在人和小鼠中,KAT2A表達的增加與關節炎癥有關
收集了健康對照、穩定型RA (SRA)患者、活動性RA (ARA)患者的PBMC樣本,并分析了KAT2AmRNA的表達情況。RA患者,尤其是ARA患者的KAT2A mRNA水平顯著高于健康對照。KAT2A mRNA水平較高的RA患者,IL1B的表達水平也相應較高。通過免疫組化(IHC)進一步分析骨關節炎(OA)和RA樣本滑膜組織中KAT2A的表達,直接證實了RA患者炎癥膝關節中KAT2A表達升高。
接下來建立了CIA模以評估小鼠滑膜組織中KAT2A表達的變化。隨著關節炎的進展,CIA模型滑膜組織樣品中KAT2A mRNA和蛋白的表達均顯著升高,與人類RA患者的現象一致。關節炎誘導后CIA模型滑膜巨噬細胞中KAT2A的mRNA表達明顯增高。此外,KAT2A的表達與關節炎小鼠滑膜巨噬細胞中Il1b的表達和臨床評分有很強的相關性。這些數據表明,無論是在RA患者還是小鼠CIA模型中,KAT2A的表達都與關節炎癥呈正相關。
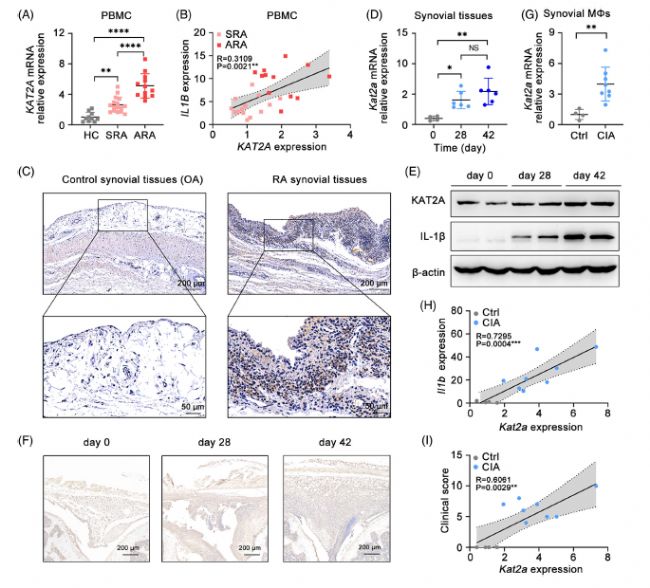
Fig 1. 在RA患者和CIA模型小鼠中,KAT2A表達升高與關節炎癥有顯著相關性
2. KAT2A抑制劑改善關節炎的炎癥病理
給CIA模型小鼠注射選擇性KAT2A酶抑制劑MB-3。MB-3顯著抑制炎癥性關節炎的發展,治療小鼠的臨床評分較低,關節腫脹較輕。microCT顯示MB-3減輕了指指骨和腕關節的骨侵蝕程度,從而改善了骨密度和骨體積。此外,膝關節組織學分析顯示,給藥MB-3后CIA模型小鼠關節囊腫脹減輕,滑膜增生減輕,膝關節炎癥細胞浸潤減少。Safranin O染色發現,KAT2A抑制也抑制了CIA小鼠膝關節軟骨的破壞。以上數據表明,抑制KAT2A可顯著減輕關節炎的病理損傷,提示KAT2A是關節炎的關鍵致病因子。
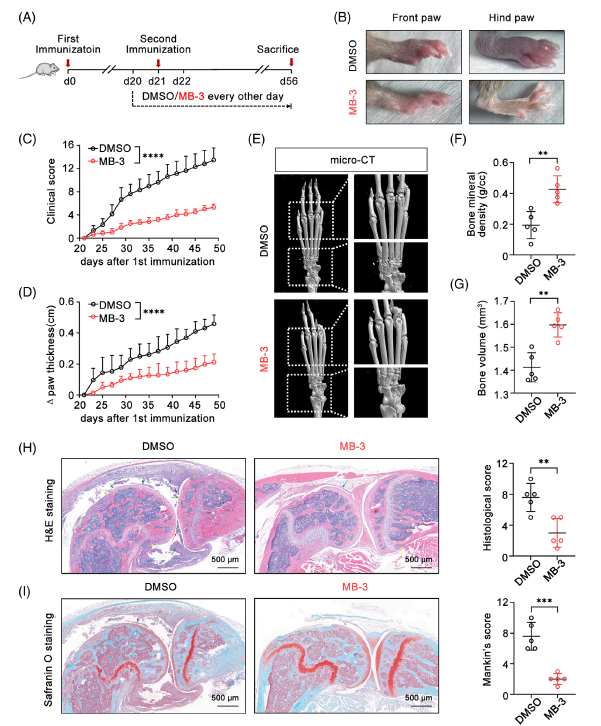
Fig 2. KAT2A抑制劑可減輕CIA模型小鼠的炎癥病理和組織損傷
3. KAT2A抑制劑抑制CIA模型小鼠炎癥和免疫紊亂
免疫失調在很大程度上決定了RA的發病機制。對照組CIA小鼠的病理性脾腫大在MB-3給藥后得到改善,這表明KAT2A抑制可能直接影響CIA模型的炎癥病。分析了血清中促炎細胞因子的分泌。給藥MB-3的CIA小鼠血清中IL-1β、IL-6和TNF-α濃度顯著降低。MB-3處理的CIA小鼠中,CD4+和CD8+高表達CD44的T細胞群減少。CIA模型小鼠發現MB-3給藥大大降低了Th1和Th17細胞的頻率。在MB-3處理小鼠的脾臟和pln發現Tregs頻率增加。KAT2A抑制顯著降低了CIA模型小鼠脾臟中 Tfh細胞的數量。KAT2A抑制通過抑制促炎細胞因子的產生和糾正隨后的T淋巴細胞失衡來改善RA的免疫損傷。

Fig 3. KAT2A抑制劑改善CIA模型小鼠的炎癥和免疫紊亂
4. KAT2A抑制劑在體內控制脂多糖(LPS)誘導的全身炎癥
通過腹腔注射LPS建立了不依賴于適應性免疫細胞的急性全身炎癥模型,LPS觸發先天免疫系統,特別是巨噬細胞的激活,包括促炎細胞因子的產生以及炎癥細胞的浸潤。MB-3預處理改善了LPS誘導的內毒素休克模型肺組織的炎癥損傷,改善了炎癥細胞浸潤和出血的病理改變,MB-3預處理導致內毒素休克模型血清細胞因子IL-1β、IL-6、TNF-α濃度大幅降低。腹腔注射LPS可觸發由炎性巨噬細胞及其分泌促炎細胞因子驅動的腹膜炎模型。通過流式細胞術分析腹膜細胞群,發現MB-3預處理抑制LPS小鼠腹腔內F4/80+CD11b+巨噬細胞的浸潤。MB-3抑制KAT2A通過調節先天免疫細胞的效應功能,在體內限制LPS誘導的全身性炎癥。
5. NLRP3炎性體啟動需要KAT2A
通過RNA-seq分析了LPS在DMSO或MB-3處理的小鼠骨髓源性巨噬細胞(BMDMs)中誘導的全局基因表達譜。KAT2A抑制導致促炎基因的大量減少。與DMSO處理的BMDM相比,MB-3處理導致RA功能基因的顯著表達差異,這與體內CIA模型的結果一致。MB-3處理介導的差異表達基因也在NLRP3炎癥小體通路中富集。KEGG通路顯示,上調基因在NF-κB信號、nod樣受體信號、細胞因子-細胞因子受體和RA中富集。
MB-3處理抑制LPS誘導的IL-1β和NLRP3基因轉錄呈劑量依賴性,免疫印跡分析也證實了這一點。MB-3處理也抑制了不同LPS刺激時間下IL-1β和NLRP3 mRNA和蛋白的表達水平。MB-3對IL-6的轉錄和分泌具有抑制作用,這也證實了RNA-seq的結果。用特異性siRNA沉默KAT2A可顯著抑制IL- 1β和NLRP3的誘導表達。為了深入研究KAT2A的功能,利用另外兩種KAT2A特異性抑制劑CPTH2和PU139來觀察它們對巨噬細胞活化的影響。這兩種抑制劑的治療都抑制了脂多糖誘導的BMDMs中Il1b和Nlrp3的轉錄,免疫印跡實驗也證實了這一點。CPTH2或PU139對KAT2A的藥理學抑制也會抑制NLRP3炎癥小體的啟動。KAT2A通過LPS刺激促進Il1b和Nlrp3的誘導表達,這對于NLRP3炎性小體的體外啟動是必不可少的。
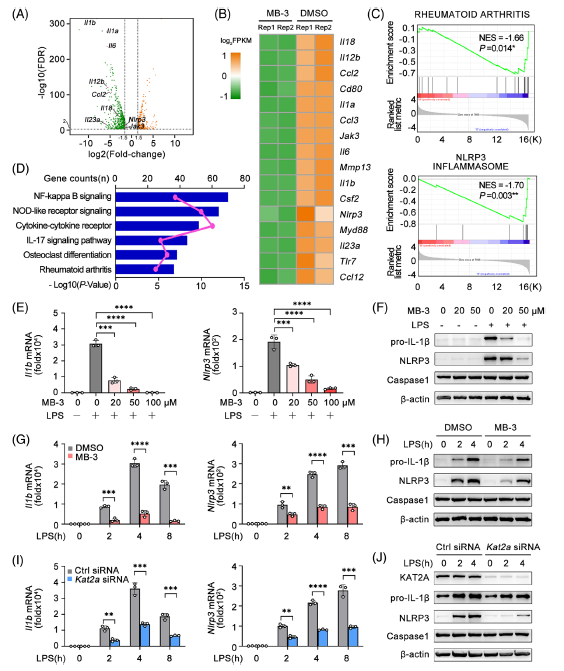
Fig 4. 在啟動階段,NLRP3炎性體的激活需要KAT2A
用三磷酸腺苷(ATP)、尼日利亞菌素(Nigericin)和尿酸鈉(MSU)等NLRP3炎癥小體激活劑處理LPS啟動的巨噬細胞,觀察KAT2A抑制劑或敲除對NLRP3炎癥小體的影響。MB-3處理有效地降低了ATP、尼日利亞菌素或MSU刺激后IL-1β的分泌,但對TNF-α的分泌沒有影響。
巨噬細胞中IL-6的分泌也被MB-3處理抑制,以響應LPS啟動和ATP、Nigericin或MSU的刺激。KAT2A沉默導致促炎細胞因子如IL-1β和IL-6的產生減少,但TNF-α的產生沒有減少。Caspase 1的裂解是炎性小體加工和分泌IL-1β的關鍵步驟。通過檢測培養巨噬細胞上清中caspase 1和IL-1β的蛋白表達,研究了KAT2A對NLRP3炎性體活化的影響。結果表明,MB-3處理可以劑量依賴性地抑制ATP或尼日利亞蛋白刺激后前il -1β和caspase 1蛋白的裂解。M B - 3處理以劑量依賴的方式顯著抑制ASC寡聚化。免疫熒光(IF)檢測顯示,尼日利亞菌素處理LPSprimed巨噬細胞可產生ASC斑點樣寡聚物結構,MB-3處理可抑制ASC斑點樣寡聚物結構的形成。
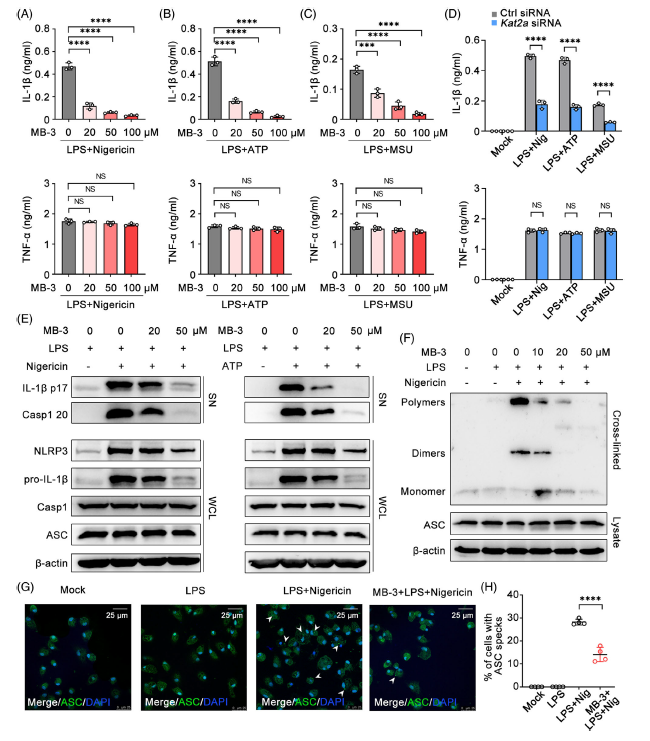
Fig 5. KAT2A促進NLRP3炎性體組裝和IL-1β加工
7. KAT2A通過抑制NRF2通路支持炎性巨噬細胞的代謝重編程
MB-3處理顯著抑制LPS刺激BMDMs的細胞ROS生成。巨噬細胞通過代謝重編程來促進巨噬細胞效應功能,以應對各種炎癥刺激。在LPS激活的巨噬細胞中,TCA明顯斷裂導致線粒體呼吸受損和糖酵解增強。LPS刺激導致ATP和乳酸的產生增加,而MB-3處理則降低了ATP和乳酸的產生,表明LPS觸發的線粒體呼吸向厭氧糖酵解的轉換受到MB-3的破壞。通過直接測定LPS刺激的巨噬細胞在處理或不處理MB-3的情況下的細胞外酸化率(ECAR)來評估細胞糖酵解水平。結果顯示,MB-3治療顯著降低了巨噬細胞的基礎和最大ECAR,證實了MB-3治療后糖酵解水平受。
為了揭示KAT2A對巨噬細胞代謝重編程的潛在機制,重新分析了MB-3或DMSO預處理的LPS活化BMDM的RNA-seq數據,發現差異表達基因在NRF2通路中富集, MB-3處理的BMDM中,許多上調基因與NRF2功能和轉錄活性相關,與GSEA結果一致。MB-3處理誘導LPS激活的BMDM中NRF2蛋白表達上調。用特異性siRNA在BMDMs中沉默NRF2的表達,然后用MB-3處理NRF2沉默的BMDMs和對照BMDMs,研究NRF2參與IL-1β的轉錄和蛋白質合成。雖然MB-3對對照BMDMs有明顯的抑制作用,但對NRF2沉默BMDMs中NLRP3炎性小體的啟動(包括NLRP3)和il -1β前蛋白的合成沒有影響。這些結果表明,MB-3對炎癥反應的抑制作用依賴于NRF2的上調及其抗炎作用。
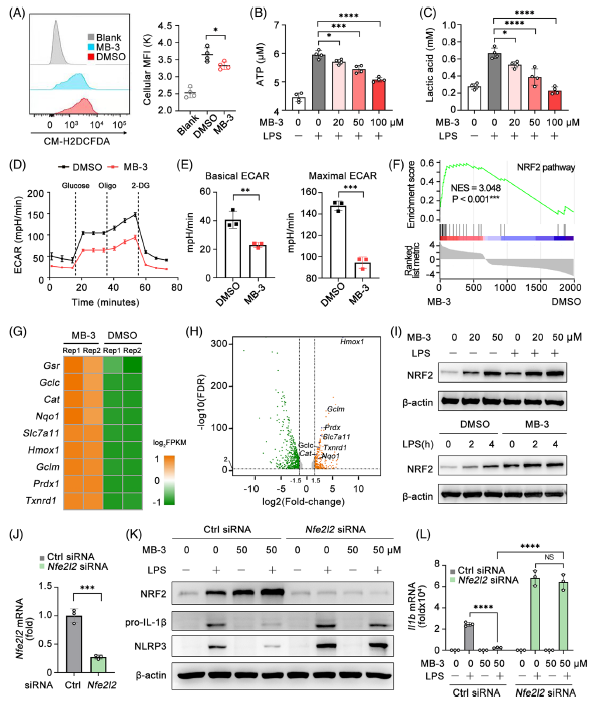
Fig 6. KAT2A介導的NRF2通路抑制允許炎癥巨噬細胞的代謝重編程
8. KAT2A協調組蛋白乙酰化與NRF2轉錄活性,用于Il1b和Nlrp3轉錄
分別轉染含有Il1b啟動子、Nlrp3啟動子或Tnf啟動子的熒光素酶報告質粒,進行了雙熒光素酶報告基因檢測。MB-3劑量依賴性地抑制traf6誘導的Il1b和Nlrp3啟動子激活,但對Tnf啟動子激活影響不大。來自公共數據庫的ChIP-seq分析顯示,在LPS刺激的巨噬細胞中,NRF2特異性結合Il1b和Nlrp3的啟動子,其中NRF2影響H3K9ac水平使用NRF2抗體或H3K9ac抗體進行ChIP-qPCR檢測。作為陽性對照,MB-3處理顯著促進了Nqo啟動子上NRF2的富集。MB-3處理也增強了NRF2對Il1b和Nlrp3啟動子的富集,表明NRF2轉錄抑制因子活性升。同時,使用H3K9ac抗體的ChIP-qPCR結果顯示,MB-3抑制KAT2A也降低了Il1b和Nlrp3啟動子上的H3K9ac水平,表明MB-3處理介導的轉錄激活能力喪失。MB-3協調了受抑制的組蛋白H3K9ac修飾和NRF2轉錄抑制因子活性的增強,以控制Il1b和Nlrp3 mRNA的表達。KAT2A通過抑制NRF2介導的代謝重編程,驅動炎性巨噬細胞NLRP3炎性小體的異常激活和IL-1β的過量產生,導致RA的進行性關節損傷。
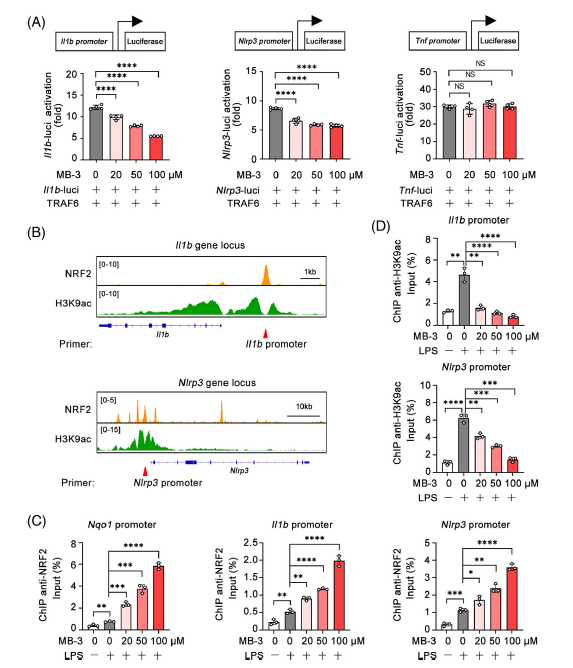
Fig 7. KAT2A協調組蛋白乙酰化和NRF2轉錄活性,促進Il1b和Nlrp3的表達
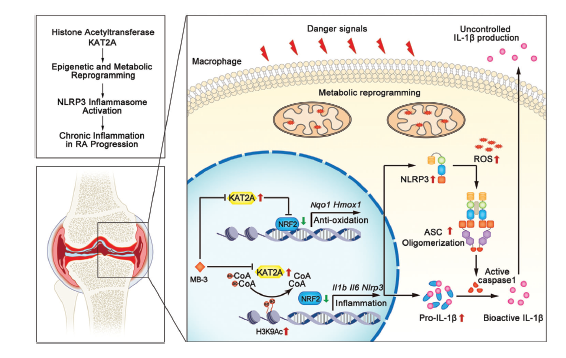
Fig 8. KAT2A在炎性巨噬細胞表觀遺傳和代謝重編程中的圖示
參考文獻:
Zhang Y, Gao Y, Ding Y, Jiang Y, Chen H, Zhan Z, Liu X. Targeting KAT2A inhibits inflammatory macrophage activation and rheumatoid arthritis through epigenetic and metabolic reprogramming. MedComm (2020). 2023 Jun 11;4(3):e306. doi: 10.1002/mco2.306IF: 9.9 . PMID: 37313329; PMCID: PMC10258526.



