
PEAKS在質譜數據鑒定多肽錯誤發現率FDR評估的應用
概覽
每個肽鑒定軟件的核心功能都是對肽和MS/MS譜的匹配質量的評估。對于數據中的每張MS/MS譜圖,軟件都會搜索蛋白質數據庫,以找到最高肽譜匹配分數的肽。譜圖與得分最高的肽之間的匹配通常稱為肽-譜匹配(peptide-spectrum match,PSM)。
一系列原因可能導致PSM錯誤,其中包括:
01.低質量的譜圖;
02.肽不在數據庫當中
03.打分體系不夠完善。為了控制結果質量,PSM按其得分來排序。通過選擇合適的分數閾值,可以得到滿足高于閾值條件質量的PSMs(圖1)。錯誤發現率FDR指的是錯誤的PSMs和在閾值之上的PSMs總數的比率。
用Target-Decoy方法來估算FDR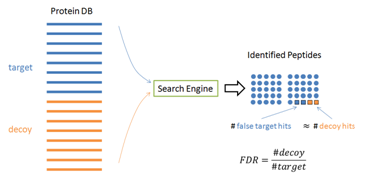
Target-Decoy方法使用的常見誤區
如果使用得當,Target-Decoy方法在統計學上是估算FDR的合理方法。不過,這種方法的誤用很常見,并且會導致對結果質量的過高評估。在這里,我們簡要總結一些常見的錯誤。需要強調的是,前面講到的“相同大小”和“均勻分布”是正確使用Target-Decoy方法的前提條件。我們即將看到,大多數Target-Decoy方法的使用錯誤都是由于違反了這些先決條件。
01錯誤1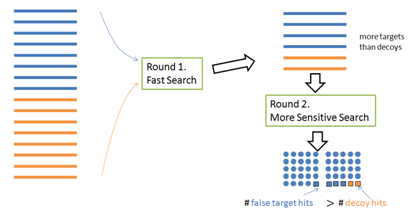
02錯誤 2
使用Target-Decoy方法時,通過蛋白鑒定信息,來反饋給搜索軟件中的肽譜匹配進行獎勵性加分。
一個蛋白的PSM越多,代表這個蛋白的置信度就越高。因此,許多軟件工具會給來自高置信度蛋白的肽加分。盡管這樣做可以提高搜索的靈敏度,卻讓Target-Decoy方法變得不準確:會出現更多具有高分的Target蛋白的匹配;因此,因高分帶來的錯誤的target蛋白匹配將會比decoy錯配獲得的蛋白更多。錯誤匹配將不會均勻分布。
03錯誤 3
通過應用Target-Decoy方法時,用重新訓練出的模型來進行對結果的重排。
這種結果重排的策略最近被越來越廣泛的使用,因為它可以提高搜索的靈敏度。然而,這也會令Target-Decoy方法變得不準確:一個較為粗放的重新學習算法會用到過多的參數,使得數據出現過度擬合并消除decoy hit(但并不是target庫中的錯配)。因此,這種策略僅適用于當重新訓練算法的設計考慮了過擬合問題,并且數據集非常大的情況。
Decoy-Fusion方法有一個簡單的改進可以避免前兩個常見錯誤——PEAKS DB的論文[1]提出了一種decoy- fusion的方法。decoy- fusion方法不是將target和decoy數據庫連接在一起,而是將同一蛋白質的target序列和decoy序列連接在一起,作為“fusion”序列(圖5)。這個簡單的更改會產生一些有意義的不同。對于兩輪搜索問題,第二輪的target和decoy長度仍然相同。對于蛋白質獎勵性得分問題,相同數量的給分將同樣的應用于同一融合序列的target和 decoy部分。 因此,“相同大小”和“均勻分布”的先決條件被重新創建;FDR值能夠被重新準確的估算。PEAKS軟件的內置結果驗證正是使用的這種decoy- fusion方法。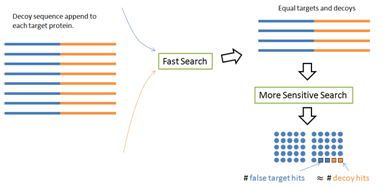
圖5:decoy- fusion方法將target和decoy序列“融合”在一起。因此,即便使用兩輪搜索算法,target序列和decoy序列也保證具有相同的長度。
參考文獻

(點擊圖片即可查看活動詳情)
如果您想深入了解更多關于PEAKS 軟件更多內容,歡迎掃描下方二維碼關注我們!
從質譜數據中鑒定多肽通過軟件實現自動化。然而,就像科學實驗需要使用對照進行一樣,軟件的多肽鑒定結果也需要經過統計驗證以避免假陽性。對于當今的肽鑒定,最被廣泛接受的結果驗證方法是錯誤發現率(FDR)。這篇文章解釋了什么是FDR;它是如何實際運算的;以及使用FDR控制中的一些常見錯誤。
每個肽鑒定軟件的核心功能都是對肽和MS/MS譜的匹配質量的評估。對于數據中的每張MS/MS譜圖,軟件都會搜索蛋白質數據庫,以找到最高肽譜匹配分數的肽。譜圖與得分最高的肽之間的匹配通常稱為肽-譜匹配(peptide-spectrum match,PSM)。
一系列原因可能導致PSM錯誤,其中包括:
01.低質量的譜圖;
02.肽不在數據庫當中
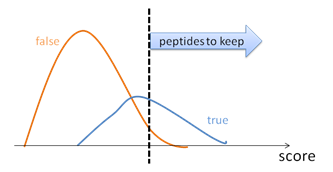
03.打分體系不夠完善。為了控制結果質量,PSM按其得分來排序。通過選擇合適的分數閾值,可以得到滿足高于閾值條件質量的PSMs(圖1)。錯誤發現率FDR指的是錯誤的PSMs和在閾值之上的PSMs總數的比率。
圖1:軟件使用評分功能來區分鑒定結果的真假。FDR是誤報高于用戶指定的分數閾值的部分。
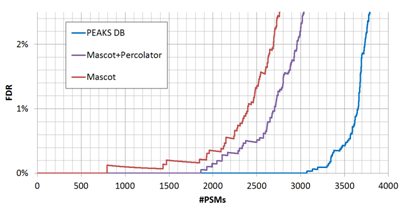
通過調整閾值,結果的準確度(FDR) 可以與靈敏度(鑒定到的數量)進行轉化。軟件不同,其評分體系不同,可能具有顯著不同的權衡效率,如圖 2中的FDR曲線所示。
圖2:不同肽鑒定軟件的性能可以通過其FDR曲線進行比較。在同一FDR閾值下,表現最好的應該是能夠鑒定到最多的PSM(數據來自圖[1])。
用Target-Decoy方法來估算FDR
在實踐當中,很難分辨哪個PSM是錯誤的—否則這些錯誤的PSMs可以被算法刪除以實現零錯誤率。 因此,Target-Decoy方法[1]在實踐中被廣泛用于估算FDR。在這個方法當中,軟件會在相同大小的target數據庫和decoy數據庫進行搜索。 如果Decoy庫建的是足夠準確的,那么軟件鑒定出的錯誤應當在目標庫和誘餌庫當中均勻分布。由于所有的decoy中得到的鑒定都應當是錯誤的,FDR 可以通過 FDR = (# Decoy hit) / (# target hit)來估計。
圖3:使用正確構建的decoy庫,錯誤匹配將均勻分布在target和decoy上。因此,decoy hit數量可用于估計FDR。
Target-Decoy方法使用的常見誤區
如果使用得當,Target-Decoy方法在統計學上是估算FDR的合理方法。不過,這種方法的誤用很常見,并且會導致對結果質量的過高評估。在這里,我們簡要總結一些常見的錯誤。需要強調的是,前面講到的“相同大小”和“均勻分布”是正確使用Target-Decoy方法的前提條件。我們即將看到,大多數Target-Decoy方法的使用錯誤都是由于違反了這些先決條件。
01錯誤1
使用方法Target-Decoy來驗證搜索軟件中的多輪搜索方法。
為了加快搜索速度,多輪搜索算法通常在第一輪從大型數據庫中選擇一個蛋白質的候選列表,然后在第二輪的蛋白質候選列表(而不是整個數據庫)中鑒定到更多的PSM。然而,這種方法使用Target-Decoy法將無效:在第一輪中選擇的目標蛋白多于Decoy蛋白數目;因此,在第二輪中,target和decoy的大小不同(圖4)。
為了加快搜索速度,多輪搜索算法通常在第一輪從大型數據庫中選擇一個蛋白質的候選列表,然后在第二輪的蛋白質候選列表(而不是整個數據庫)中鑒定到更多的PSM。然而,這種方法使用Target-Decoy法將無效:在第一輪中選擇的目標蛋白多于Decoy蛋白數目;因此,在第二輪中,target和decoy的大小不同(圖4)。
圖4:第一輪中保留了更多的target蛋白。因此,在target蛋白庫中將鑒定到更多的隨機錯誤。所以說,Decoy hit次數不能再被用于估算錯誤匹配數。
02錯誤 2
使用Target-Decoy方法時,通過蛋白鑒定信息,來反饋給搜索軟件中的肽譜匹配進行獎勵性加分。
一個蛋白的PSM越多,代表這個蛋白的置信度就越高。因此,許多軟件工具會給來自高置信度蛋白的肽加分。盡管這樣做可以提高搜索的靈敏度,卻讓Target-Decoy方法變得不準確:會出現更多具有高分的Target蛋白的匹配;因此,因高分帶來的錯誤的target蛋白匹配將會比decoy錯配獲得的蛋白更多。錯誤匹配將不會均勻分布。
03錯誤 3
通過應用Target-Decoy方法時,用重新訓練出的模型來進行對結果的重排。
這種結果重排的策略最近被越來越廣泛的使用,因為它可以提高搜索的靈敏度。然而,這也會令Target-Decoy方法變得不準確:一個較為粗放的重新學習算法會用到過多的參數,使得數據出現過度擬合并消除decoy hit(但并不是target庫中的錯配)。因此,這種策略僅適用于當重新訓練算法的設計考慮了過擬合問題,并且數據集非常大的情況。
Decoy-Fusion方法有一個簡單的改進可以避免前兩個常見錯誤——PEAKS DB的論文[1]提出了一種decoy- fusion的方法。decoy- fusion方法不是將target和decoy數據庫連接在一起,而是將同一蛋白質的target序列和decoy序列連接在一起,作為“fusion”序列(圖5)。這個簡單的更改會產生一些有意義的不同。對于兩輪搜索問題,第二輪的target和decoy長度仍然相同。對于蛋白質獎勵性得分問題,相同數量的給分將同樣的應用于同一融合序列的target和 decoy部分。 因此,“相同大小”和“均勻分布”的先決條件被重新創建;FDR值能夠被重新準確的估算。PEAKS軟件的內置結果驗證正是使用的這種decoy- fusion方法。
圖5:decoy- fusion方法將target和decoy序列“融合”在一起。因此,即便使用兩輪搜索算法,target序列和decoy序列也保證具有相同的長度。
參考文獻
- Zhang J, Xin L, Shan B, Chen W, Xie M, Yuen D, Zhang W, Zhang Z, Lajoie G.A., Ma B, PEAKS DB: De Novo Sequencing Assisted Database Search for Sensitive and Accurate Peptide Identification. Mol. Cell. Proteomics. 11, M111.010587 (2012).
- Xin, L., Qiao, R., Chen, X. et al. A streamlined platform for analyzing tera-scale DDA and DIA mass spectrometry data enables highly sensitive immunopeptidomics. Nat Commun 13, 3108 (2022). doi:10.1038/s41467-022-30867-7
(點擊圖片即可查看活動詳情)
如果您想深入了解更多關于PEAKS 軟件更多內容,歡迎掃描下方二維碼關注我們!




Copyright(C) 1998-2025 生物器材網 電話:021-64166852;13621656896 E-mail:info@bio-equip.com