
8種表觀轉錄組m6A MeRIP-seq的差異甲基化區(qū)域(DMR)分析軟件比較
RNA甲基化是近年來研究基因表達調控轉錄后變化的重要研究領域,包括N6-甲基腺苷(m6A)在內的各種類型RNA甲基化參與人類疾病發(fā)展。MeRIP-seq作為一種新興的在轉錄組范圍內定量檢測m6A水平的測序技術,拓展了RNA表觀遺傳學研究的基礎和臨床應用,且呈上升趨勢。RNA甲基化數據分析的基本問題之一是通過對比病例和對照來鑒定差異甲基化區(qū)域(DMR)。現有開發(fā)了多種用于DMR檢測的分析方法,但缺乏對這些分析方法的綜合評估。
本文利用模擬數據和真實數據,全面評估了DMR calling的所有8種現有方法:FET-HMM、exomePeak(2.16.0版)、MeTDiff(1.1.0版)、DRME、QNB(1.0版)、exomePeak2(1.9.1版)、RADAR(0.2.4版)和TRESS(1.4.0版)。
模擬分析采用Gamma–Poisson模型和logit線性框架,并調試適應各種樣本量和DMR比例進行基準檢測。所有8種方法在低input水平區(qū)域中觀察到低靈敏度,但樣本量增加會大大提高靈敏度。TRESS和exomePeak2在檢測精確度、FDR(False DiscoveryRate)、I型錯誤(type-I error)調控和運行時間等指標上表現最好,但受限于低靈敏度。DRME和exomePeak以FDR(假發(fā)現率)和I型錯誤(type-I error)膨脹為代價獲得高靈敏度。三個真實數據集分析表明,這些方法在鑒定DMR長度和唯一發(fā)現區(qū)域有不同偏好性。
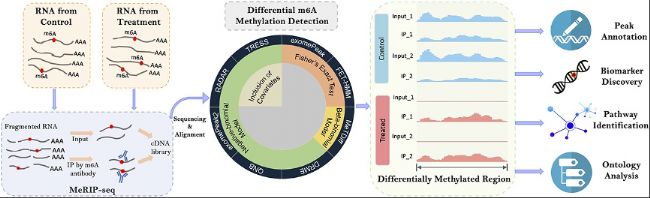 圖1:MeRIP-seq實驗和DMR檢測示意圖
圖1:MeRIP-seq實驗和DMR檢測示意圖
MeRIP-seq從RNA樣本生成配對的IP數據和input對照數據。將測序reads比對至參考基因組,然后通過最近開發(fā)的統計方法鑒定差異甲基化區(qū)域(DMR)。其核心統計模型和特征列于餅圖內圓,下游對DMR基因進行peak注釋、biomarker發(fā)現、通路(pathway)鑒定和基因本體(GO)分析。

表1:現有差異RNA甲基化分析方法,方法按時間順序排列。
TDR:真發(fā)現率(True Discovery Rate),即在某個截止點前被鑒定區(qū)域中所占的真陽性。
FDR:假發(fā)現率(False Discovery Rate)。
隨著MeRIP-seq在過去幾年的廣泛使用,已經開發(fā)了幾種檢測DMR的計算方法,所有已知8種方法的詳細信息見表1(按開發(fā)時間順序排列)。作為第一個發(fā)布的工具,exomePeak在兩種實驗條件下對input對照和IP樣本的歸一化reads數應用Fisher精確檢驗(Fisher’s exact test,FET),因為使用所有重復的總reads,它忽略了生物學重復之間的異質性。隨后改進exomePeak開發(fā)了MeTDiff和FET-HMM方法,MeTDiff假設β二項分布,并通過似然比檢驗(Likelihood Ratio Test,LRT)比較不同條件下的甲基化水平,然而MeTDiff沒有很好解決測序深度的技術差異;FET-HMM采用了FET的改進版本,并使用FET的二值決策作為差異甲基化狀態(tài)的觀察,隨后在檢測到的甲基化區(qū)域內的小箱子上擬合隱馬爾可夫模型(Hidden Markov Model,HMM),以合并沿基因組的依賴性,然而FET-HMM在合并每組中的重復檢測,忽略了生物學重復之間的組內變化。2016年開發(fā)的DRME解決了這個問題,尤其是在小樣本量的情況下,DRME假設IP和input對照計數數據均為負二項式模型,且僅使用input對照數據來預測背景基因表達,通過計算基于IP數據統計顯著性來檢測DMR。DRME的作者后來改進了他們的模型并開發(fā)了QNB,QNB也使用負二項式模型,與DRME不同的是,QNB在背景表達式的預測和檢測統計的計算中結合了input對照和IP數據,DRME和QNB的共同限制是,兩者都將ip內和input內的變化作為目標變化,但在MeRIP-seq中信號為IP/input比,應該嚴格建模該比方差。后來exomePeak的作者又提出了exomePeak2,與exomePeak相比,exomePeak2解釋了IP效率和GC含量偏差變化,當存在多個重復時,exomePeak2 calling DESeq2通過將IP和input視為成對樣本來鑒定DMR。
上述6種DMR分析方法僅適用于兩組間的比較。在真實的生物學實驗中,特別是在大型研究中,經常出現混雜協變量(如年齡或性別),但在上述方法中無法正確解釋。為了解決這個問題,最近提出了兩種方法:RADAR和TRESS,兩種方法都使用線性框架將甲基化水平與實驗因子相關聯。RADAR采用泊松隨機效應模型(Poisson random effect model),而TRESS采用伽馬-泊松分布(Gamma–Poisson distribution)。TRESS與RADAR在兩個方面不同,第一個區(qū)別是TRESS假設原始reads數遵循負二項式分布,通常用于建模各種測序數據類型。而RADAR假設預處理(從文庫大小歸一化開始,然后進行input對照調整)的計數數據遵循泊松分布,預處理后的數據不再是計數格式,因此泊松假設模棱兩可。另一個區(qū)別在于一旦模型擬合,TRESS可以檢測所有包含因子或其中任何線性組合的影響;而使用RADAR檢測不同因子,需要重新提供設計矩陣并再次擬合模型,在計算上比較低效。總的來說,上面描述的方法列在表1中,顯示了input數據類型、算法簡要描述和使用中的各自優(yōu)缺點。
數據生成模型和模擬(DATA GENERATIVE MODEL AND SIMULATION)
模擬框架的核心是伽瑪-泊松分布,并為適應MeRIP-seq數據進行了適當修改。假設總共有10000個候選DMR,其中10%在處理和未處理的條件下差異甲基化。每種條件下的重復次數從2到10不等,以評估樣本量對DMR calling的影響。
整體差異peaks比較(OVERALL DIFFERENTIAL PEAK COMPARISON)
對8種m6A DMR檢測方法進行基準檢測,每種場景下進行20次模擬。在一個共同實驗設計下評估所有方法,作為整體比較基線,分別研究每個影響因子。
在每次模擬中,10000個候選DMR中的1000個被設計為真DMR,在病例組和對照組中都有三個重復。使用幾個評估指標來評估八種DMR檢測算法的性能,如使用真發(fā)現率(TDR)來分析生物標志物發(fā)現的精確度。還研究了ROC曲線(Receiver Operating Characteristic curve)、靈敏度和假發(fā)現率等經典指標。值得注意的是,FETHMM共有三種策略:“FHB”、“FHC”和“FastFHC”。本文使用FHC而非默認設置(FastFHC),因為默認設置中的編碼有對P值的異常操作。
圖2顯示了在上述基線模擬場景中所有方法之間的DMR calling性能比較。如圖2A所示,所有方法的TDR中,每個截止點,TRESS和exomePeak2的TDR值是最高的,且?guī)缀跸嗤?/strong>。exomePeak、FETHMM、DRME和QNB生成的TDR相當,但略低于TRESS和exomePeak2。圖2B顯示TRESS、exomePeak2、exomePeak、FETHMM和DRME的AUC最高,而MeTDiff的AUC最低。值得注意的是,盡管TDR和ROC在方法比較中都是有效的指標,但TDR更具信息性,因為排名靠前的指標與生物標志物發(fā)現更相關。圖2C中TRESS、exomePeak2、exomePeak、FETHMM和DRME的P值高度相關(Spearman相關性≥0.93),而MeTDiff得到的P值與其他方法差異較大。分析了最高和最低相關性的四對方法,并證明了FETHMM、exomePeak和exomePeak2之間的相似性。在圖2D、E中,Benjamini–Hochberg調整后的P值計算靈敏度和假發(fā)現率,以0.05為截止值。盡管DRME、FETHMM和exomePeak具有高靈敏度,但其FDR值也很高。結合這兩個指標,表明DRME、FETHMM和exomePeak的I型錯誤(type-I error)膨脹。MeTDiff和QNB在模擬中表現出不穩(wěn)定性。MeTDiff很難鑒定出真陽性,因此并不是在所有的比較中都表現良好。TRESS和exomePeak2實現了幾乎相同的最佳整體性能,在保持低FDR的同時發(fā)現了相當一部分真陽性。圖2F中還總結了使用平均靈敏度和FDR的聯合分布的總體性能。理想情況下,好的方法應該具有高靈敏度,同時保持低FDR,因此位于左上角區(qū)域的方法是首選方法。
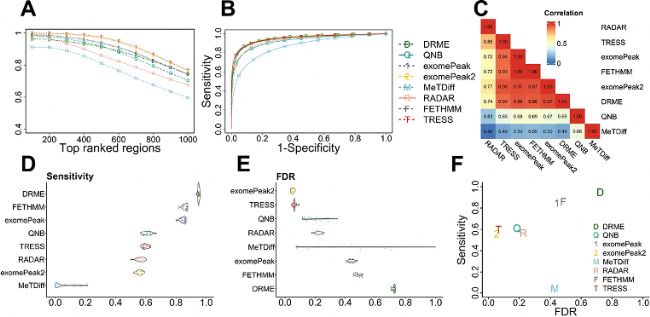
圖2:m6A-DMR檢測方法的性能比較。
- 每種方法鑒定的排名靠前區(qū)域的真發(fā)現率(TDR)。TDR定義為調整后p值排名前靠前區(qū)域中真DMR占比。
- DMR檢測方法的受試者工作特征(ROC)曲線。
- 8種方法的p值相關性熱圖。
D-E. 每種方法的靈敏度和FDR分布的小提琴圖,用BH調整后的p值計算。
F. 每種方法檢測DMR的平均靈敏度與FDR。模擬在三個病例組、三個對照組、10%真DMR的情況下進行。N=20次模擬。
樣本量(SAMPLE SIZE)
接下來研究樣本量對DMR calling精確度的影響,因為樣本量通常是實驗設計中的主要參數。本研究兩組的模擬樣本量分別為2、3、5、7和10,每種條件下2、3、7、10個樣品的TDR分別如圖3A-D所示。幾乎所有方法在靠前排名 (如前100或前200)calling區(qū)域獲得高TDR(>0.8),且在排名靠后時顯示出精確性下降。具體來說,TRESS和exomePeak2在所有截止點上都保持最高的精確度,而MeTDiff表現最差,沿秩遞減的精確度最低。隨著樣本量增加,所有方法的精確度都有所提高。當N=7和10時,這種趨勢尤其明顯,其中方法報告的TDR值相似。在圖3E中,TDR以熱圖的形式呈現,包括所有模擬場景下的結果(N=2、3、5、7、10),按排名前400、700、1000和1500區(qū)域進行分層。總體而言,所有方法中TDR值隨區(qū)域排名提高和樣本量增加而增加。大樣本量可以大大提高檢測精確度,即使是排名中等區(qū)域(如前1000名)。RADAR和MeTDiff在小樣本量中 (N=2和3)的檢測精確度較低,但隨著樣本量增加,其性能幾乎相同。即使在極小的樣本量下(N= 2), TRESS和exomePeak2的TDR也大于0.8。在經驗貝葉斯框架(empirical Bayes framework)下,TRESS和exomePeak2在全基因組中實現了信息借用,因此其在小樣本量中的表現優(yōu)于其他方法。在其他基因組學研究中,這種建模技術已被證明是有效的統計框架,特別是對于小樣本量。總之,對于小樣本量的項目,TRESS和exomePeak2是首選。
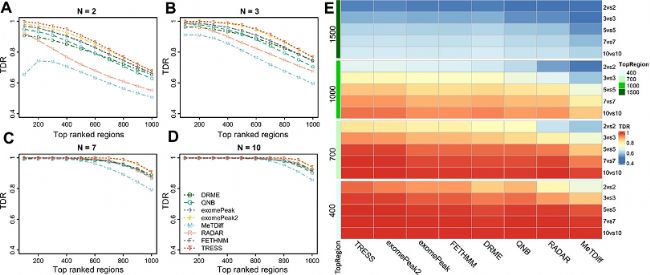 圖3:不同樣本量的DMR檢測精確度比較。
圖3:不同樣本量的DMR檢測精確度比較。
A-D. 每組中進行2、3、7和10次重復的樣本量下,每種方法鑒定的排名靠前區(qū)域的真發(fā)現率(TDR)。
E. 不同樣本量和TOP區(qū)域截止值組合下的TDR值熱圖。樣本量標注在右側,每組2個、3個、5個、7個和10個。排名靠前區(qū)域截止線標注在左側,范圍從前400名、前700名、前1000名到前1500名。方法在熱圖中按列排序。在10%真DMR下進行了N=20次模擬,取平均TDR值。
分層評估(STRATIFIED ASSESSMENT)
高通量測序數據(如批量RNA-seq)的差異表達分析準確性高度依賴于表達水平,因此本研究按input范圍分層檢測DMR準確性。根據input對照分布,候選區(qū)域根據其平均input計數分為五層:第一層1(0~10)、第2層(10~20)、第3層(20~40)、第4層(40~80)和第5層(80~+∞). 以0.05值為標稱值顯著性水,所有方法在5個分層中的靈敏度和FDR如圖4所示。方法按各層的平均值排序,當從較低分層轉到較高分層時,所有方法都提高了靈敏度(圖4A–C),低input區(qū)域通常容易受模擬噪聲影響。即使在第一層,DRME也具有較高靈敏度(>0.75),且在所有區(qū)域中具有相對較好性能。隨著樣本量增加,DRME靈敏度仍在提高。隨著樣本量增加,所有方法都表現出增加和減少的可變靈敏度,且這種性能增益對于較低層非常顯著,表明大樣本量有助于更可靠預測,尤其是對于受高背景噪聲影響更大的區(qū)域。其中,exomePeak2從樣本量的增加中受益最大,從第七位上升到第四位。對于FDR的結果,更大樣本量不如靈敏度(圖4D–F)。TRESS和exomePeak2在所有分層和樣本量大小中顯示出較小且最一致的假發(fā)現率(FDR)。在小樣本量下(N=3),MeTDiff在較低input區(qū)域的FDR較差,而隨著樣本量增加,FDR得到很大的提升。exomePeak、FETHMM和DRME受較差FDR影響,即使在大樣本情況下也是如此(N=10)。
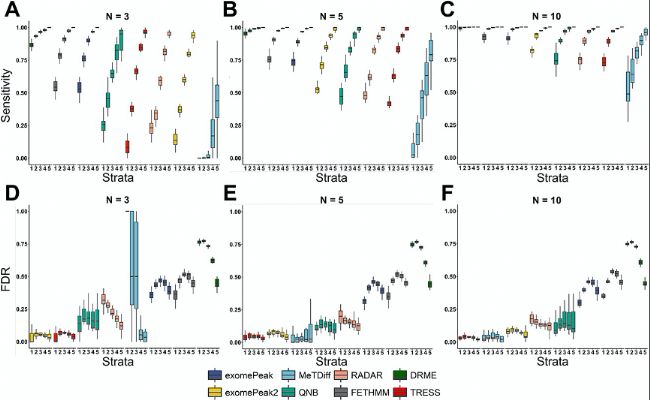 圖4:按平均input計數值分層分析靈敏度和FDR。靈敏度和FDR以BH調整后P值計算,以0.05為截止值確定顯著性。
圖4:按平均input計數值分層分析靈敏度和FDR。靈敏度和FDR以BH調整后P值計算,以0.05為截止值確定顯著性。
A–C. 分層靈敏度,每組分別設置3個、5個和10個重復。
D–F. 分層FDR,每組分別設置3個、5個和10個重復。在10%DMR下進行N=20模擬。
I型錯誤和p值有效性(TYPE I ERROR AND VALIDITY OF P-VALUES)
為了研究8種方法的I型錯誤和p值有效性,在null條件下進行假設模擬,其中沒有(0%)候選區(qū)域存在差異甲基化。使用bh調整后的p值以0.05標稱值為顯著性水平獲得每種方法鑒定的DMR。在每組設置2、3、5、7和10個重復的情況下,計算經驗I型錯誤率(表2)。在所有情況下,TRESS和FETHMM的I型錯誤率都接近0.05,表明其I型錯誤率接近標稱值。exomePeak2更為“保守”,小樣本下(N = 2、3)以低靈敏度獲得最佳FDR(圖2D、E)。DRME是最“自由”方法,與其高靈敏度和FDR相匹配(圖2D、E)。
P值有效性分析檢測了null條件下P值是否均勻分布在0和1之間,并在圖5中說明了每組使用三個重復的結果。在QQ圖(Quantile-quantile plot)中,TRESS和exomePeak2產生的p值與預期值最為一致(圖5A,位于或接近對角線參考線)。大多數方法生成自由p值(右下方區(qū)域),而FETHMM在大多數區(qū)域過于保守(左上方區(qū)域)。由于小p值在DMR檢測中更具信息性,本研究還應用-log10轉換,重點關注圖5B中小p值分布。TRESS、exomePeak2和RADAR表現最好,而其他方法提供的p值過小,表明I型錯誤膨脹。且對樣本量不敏感(圖2E),其中TRESS、exomePeak2和RADAR產生的的FDR控制得最好,也最穩(wěn)定。
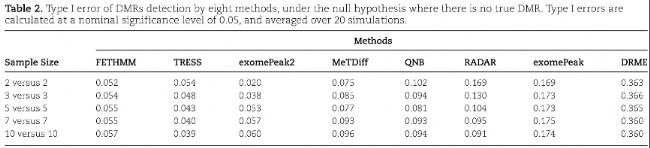 表2:在無真DMR的null假設下,八種方法檢測DMR的I型錯誤(0.05標稱值顯著性水平計算,并在20次模擬中取平均值)
表2:在無真DMR的null假設下,八種方法檢測DMR的I型錯誤(0.05標稱值顯著性水平計算,并在20次模擬中取平均值)
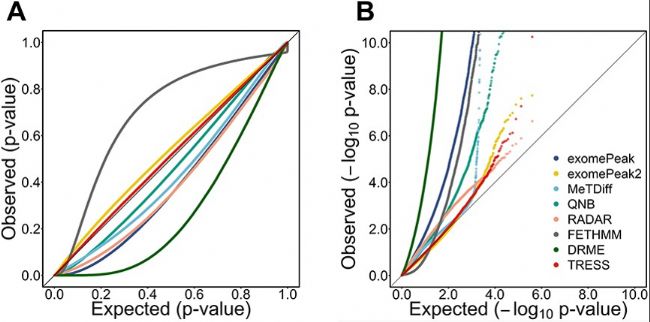 圖5:分析null條件下模擬觀察到的p值有效性。
圖5:分析null條件下模擬觀察到的p值有效性。
- QQ圖(Quantile–quantile plot)將p值分布與null下的期望分布U(0,1)進行比較。
- QQ圖進行−log10轉換,重點關注小P值。在無DMR的null假設下進行20次模擬。樣本量N=3 /組。
運行時長和內存消耗(RUNTIME AND MEMORY CONSUMPTION)
BAM文件為默認input評估每種方法的軟件運行時間和計算內存消耗。基于同一節(jié)點、同一內核和200 GB內存的高性能計算(HPC),在不同樣本量下,五種方法的運行時長如圖6A所示。隨著樣本量增加,所有方法都顯示出更長的運行時間。與其他方法相比,TRESS和exomePeak2的運行時長都更短,且隨著樣本量增加更為明顯。exomePeak和MeTDiff在所有樣本量中具有相似的運行時間。RADAR的運行時間最慢。由于大多數方法都將BAM文件作為標準input,因此進一步對計算內存消耗進行了基準檢測(圖6B)。MeTDiff和exomePeak消耗內存最少(分別為3.81 GB和4.62GB)。TRESS消耗的內存略多于MeTDiff和exomePeak。exomePeak2利用了最多的內存(170.28GB)。模擬在HPC中進行,每個方法calling都有1個節(jié)點、40個內核和200 GB可用內存。
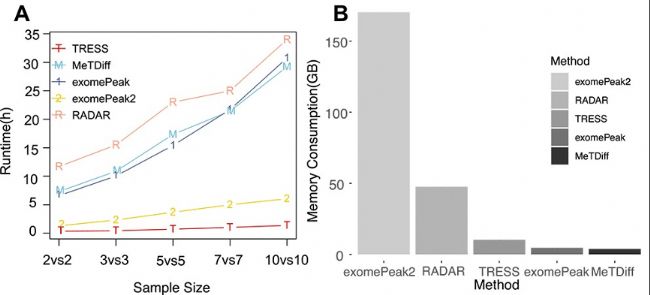 圖6:m6A DMR檢測方法的運行時長和內存消耗比較。
圖6:m6A DMR檢測方法的運行時長和內存消耗比較。
- 五種不同方法在不同樣本量下的運行時長比較,以小時為單位。
- 計算五種不同方法的內存消耗,單位為GB。
真實數據分析(REAL DATA ANALYSIS)
首先從一項研究METTL3-METTL14復合體介導哺乳動物核RNA m6A甲基化的研究中獲得了真實數據集(GSE46705),將其標記為“RD1”。在該研究中,人類HeLa細胞系有四種樣品類型:一種野生型(WT)樣品和三種處理過的樣品,這些處理對應于復合體METTL3、METLL14和WTAP的敲除(KD)。每個樣品2個重復。將TRESS、exomePeak、exomePeak2、MeTDiff和RADAR方法應用于該真實數據,以鑒定m6A差異甲基化。同時還采用了適用于分析MeRIP-seq數據的MACS3方法。MACS3已被先前的幾項研究表明其作為MeRIP-seq數據差異分析的有效工具的潛力。該研究只分析以BAM文件為input比較的方法,因此排除了QNB和DRME等以reads計數矩陣為input的方法。
原始FASTQ文件被比對到人類參考基因組hg18,使用帶有默認參數的STAR標準流程。比對后的BAM文件作為所有五種方法的input進行比較,主要分析WT和METTL3樣品之間的差異甲基化。DMR calling在FDR<0.05的顯著性水平上進行。在過濾掉短(寬度<150)和重疊區(qū)域后,TRESS、exomePeak、exomePeak2、MeTDiff和RADAR分別鑒定出1413、1397、5272、161和2924個DMR。exomePeak2鑒定出最多的DMR,而MeTDiff鑒定的DMR最少。
五種方法分析WT組與METTL3組真實數據的性能比較如圖7所示。使用ChIPseeker對DMR進行注釋(圖7A)。結果顯示,除了RADAR以外的大多數方法都支持3'UTR的DMR。RADAR偏好基因下游外顯子區(qū)(即非第一外顯子)。所有方法的啟動子和下游外顯子區(qū)均表現出相當數量的組成基因組表征。圖7B顯示了五種方法的5個重疊區(qū)域。exomePeak2發(fā)現3348個特異性DMR,是所有DMR中最高的。兩種方法之間重疊區(qū)域的最高數量是由exomePeak和exomePeak2 calling的1038個重疊,而兩種方法間重疊區(qū)域的最少數量是由TRESS和MeTDiff calling的15個重疊。DMR的peaks寬分布(log scale)如圖7C所示。TRESS偏好150–400bp中長區(qū)域,RADAR具有雙峰分布(bimodal distribution),覆蓋中長和長兩個區(qū)域。鑒定出1038個共有區(qū)域的FDR(圖7D)。與exomePeak2相比,exomePeak是一種更保守的方法。同時由TRESS、exomePeak、exomePeak2、MeTDiff和RADAR方法顯示了WT和METTL3樣品之間共有DMR的兩個示例(圖7E),這兩個區(qū)域覆蓋蛋白編碼基因TEX264(chr3)、PRICKLE4、TOMM6和USP49(chr6)。先前的研究表明,TEX264能夠激活信號受體活性,并參與蛋白-DNA共價交聯修復。USP46通過剪接體參與半胱氨酸型內肽酶活性、組蛋白H2B保守的C-末端賴氨酸去泛素化和mRNA剪接。對exomePeak2和RADAR進行GO(Gene Ontology)通路分析(圖7F),在exomePeak2的DMR中,前三個GO富集是“生長因子受體和第二信使的信號通路疾病(Diseases of signal transduction by growth factor receptors and second messengers)”、“TP53轉錄調控(Transcriptional regulation by TP53)”和“I類MHC介導的抗原處理和呈遞(Class I MHC mediated antigen processing & presentation)”。
同時在另外兩個真實數據集(GSE94613和GSE115105)中進行檢測,并將它們標記為“RD2”和“RD3”,其中, “RD2”包括12個METTL3敲低細胞系和對照的人類樣本,“RD3”包括兩個Ythdf1敲低和對照的野生型小鼠骨髓來源樹突狀細胞(BMDC)。對這兩個數據集進行相同的分析,根據DMR數量和3’UTR在三個真實數據集中的百分比對五種方法進行排序(圖7G)。exomePeak2軟件calling了三個真實數據集中最多的DMR,其次是RADAR。在3’UTR方面,除了exomePeak2和MeTDiff之間的微小差異外,數據集之間再次觀察到一致結果(圖7H)。
 圖7:真實數據的差異m6A甲基化方法。
圖7:真實數據的差異m6A甲基化方法。
- 條形圖顯示在已鑒定的DMR中各種基因組特征分布。TRESS、exomePeak、exomePeak2、MeTDiff、RADAR和MACS3采用相同的FDR 0.05截止值來calling顯著性。
- 維恩圖顯示通過五種方法鑒定的DMR重疊。
- 六種方法的峰寬分布密度圖(log scale)。
- exomePeak和exomePeak2的1038共有區(qū)域的成對FDR值散點圖。
- 共有DMR的peaks差異分析可視化的兩個例子。所有差異peaks分析均在野生型(WT)組和METTL3組之間。
- exomePeak2的DMR基因的GO分析,顯示DMR數最多。
- 三個真實數據集中DMR計數排序。
- 三個真實數據集中3’UTR百分比排序。
比較要點(Key Points)
- 新型表觀轉錄組測序技術能夠使用數據驅動的方法評估RNA修飾。
- 差異表觀轉錄組分析需要對成對的input對照和IP樣本進行適當的建模,以適應技術和生物噪聲、peaks值檢測并解決小樣本量問題。
- TRESS和exomePeak2在基準研究中表現出高TDR、低FDR和超高靈敏度。
- 檢測精確度可能會受低input表達影響,但受益于樣本量增加。
- RADAR、TRESS和exomePeak2顯示了頂級嚴格的I型錯誤控制和null下的有效p值分布。MeTDiff計算內存消耗最少,TRESS運行時間最快。
參考文獻:
Duan D, Tang W, Wang R, Guo Z, Feng H. Evaluation of epitranscriptome-wide N6-methyladenosine differential analysis methods. Brief Bioinform. 2023 May 19;24(3) pii: 7111718.



