
營養不良的心肌細胞中,組織硬度增加會觸發收縮功能障礙和端粒縮短
杜氏肌營養不良(DMD)是一種罕見的X連鎖隱性疾病,與嚴重的進行性肌肉退化有關,最終因心肺衰竭而死亡。有研究在DMD小鼠模型的心肌細胞中觀察到了增殖非依賴性端粒縮短。Alex C.Y. Chang等使用人誘導多能干細胞分化的心肌細胞(hiPSC-CMs),結合HCells高通量單細胞功能檢測系統,發現DMD hiPSC-CMs在纖維化樣生物工程水凝膠上顯示出力產生缺陷、鈣處理異常以及活性氧水平增加。此外還觀察到DMD hiPSC-CMs有絲分裂后端粒的逐漸縮短與shelterin復合物、端粒帽蛋白的下調和p53 DNA損傷應答的激活同時發生。這種端粒縮短可被抑制DMD心肌細胞收縮的(布雷他汀)blebbistatin阻斷。研究強調了纖維化硬化在DMD心肌病病因中的作用,得到數據表明端粒縮短是漸進的、收縮依賴的,并且機械力敏感,為治療干預提供了見解。文章以“Increased tissue stiffness triggers contractile dysfunction and telomere shorteningin dystrophic cardiomyocytes”為題發表于Stem Cell Reports。
背景
杜氏肌營養不良癥(DMD)是由編碼dystrophin(一種連接細胞骨架和細胞外基質的蛋白)的基因中,> 200個突變引起的,發病率為1:5,000,DMD患者表現出早期和進行性骨骼肌退化和無力,通常在30歲之前死于擴張型心肌病和呼吸衰竭。DMD在心臟中的表現為心電圖異常、收縮和舒張功能障礙、纖維化,最終導致心力衰竭。AAV介導的基因治療和基因編輯策略在動物模型中顯示出巨大前景。但因為缺乏對心力衰竭機制的理解,DMD患者接受的還是非特異性心力衰竭治療,如β受體阻滯劑或ACE抑制劑。
以前發現dystrophin缺乏與較短的端粒有關,端粒是六核苷酸TTAGGG重復序列,覆蓋并保護染色體末端。通過用mdx4cv和TERC敲除(mTR)小鼠繁育出的mdx4cv/mTRG2小鼠可如實再現擴張型心肌病表型。定量熒光原位雜交(Q-FISH)測得mdx4cv/mTRG2小鼠和DMD患者心臟組織中,心肌細胞的端粒與mdx4cv/mTRHet小鼠對照或同齡人對照的端粒相比,信號減少了約50%。但這種與增殖無關的端粒縮短的觸發機制仍然未知。
為深入了解端粒縮短的分子和細胞病因,研究人員使用由患者分化來的心肌細胞和同基因對照人類誘導多能干細胞(hiPSC-CMs),構建了培養條件下的人DMD心肌病模型,并繪制疾病進展圖,這是一種用于研究心臟缺陷的強大系統。重要的是,建立了一個生物工程平臺,允許心肌細胞以1:7的縱橫比定向生長并模擬DMD纖維化心臟的硬度。發現DMD hiPSC-CMs表現出異常的鈣處理、收縮缺陷和與增殖無關的端粒縮短。這種端粒縮短發生于有絲分裂后,是進行性的,且受收縮影響。通過在模擬健康和纖維化心肌硬度的生物工程平臺上培養hiPSC-CMs,發現DMD心臟的心肌硬度特征會加劇收縮功能障礙。
結果
01-杜氏hiPSC-CMs可再現擴張型心肌病的致病特征
為研究DMD hiPSC-CMs端粒縮短的分子基礎,將6個DMD和6個對照hiPSC-CMs細胞系分化為跳動的心肌細胞。使用了來自四個不同實驗室的來源于不同細胞類型的hiPSC細胞系,包括兩個通過CRISPR-Cas9(Con1和2對應DMD1和2)糾正dystrophin突變的同基因對,一個通過CRISPR-Cas9(Con3對應DMD3)將前六個外顯子的缺失引入健康細胞的同基因對,三個非家族健康對照(Con 4,5和6),和三個DMD細胞系(DMD4,5和6)。免疫熒光染色測定hiPSC的多能性標記物OCT4、NANOG和SOX2(圖1B),跳動的心肌細胞確定表達標志蛋白、心肌肌鈣蛋白(cTnT)和α-actinin(圖1C)。免疫熒光(圖1D)和qRT-OCR(圖1E)確定dystrophin的存在與否。
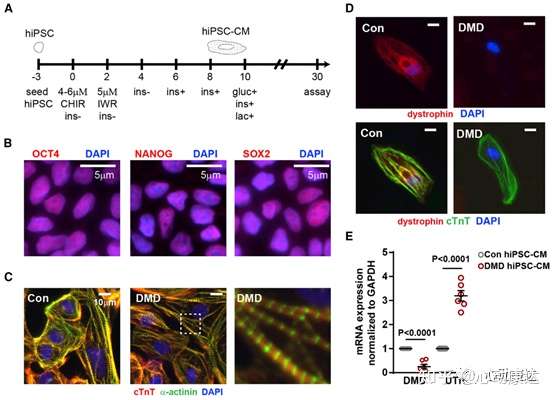
圖1 DMD hiPSC-CMs的產生。(A)hiPSC分化產生hiPSC-CMs的步驟。(B)多能干細胞標志物染色OCT4、NANOG、SOX2。(C)心臟troponin T、α-actinin、DAPI染色。(D)健康和和DMD hiPSC-CMs的dystrophin、心臟troponin T、DAPI染色。(E)hiPSC-CMs中內源性dystrophin和utrophin (UTR)表達水平。
1 Hz起搏條件下用Fura2測量以評估基礎鈣水平。觀察到靜息鈣比率變化增加(圖2A)、峰值鈣比率下降趨勢(圖2B)、瞬時振幅下降(圖2C)、除Con2/DMD2外到達峰值的時間無差異(圖2D)、90%鈣瞬變持續時間(圖2E)、衰減τ增加(圖2F)。與之前使用Fluo-4方法觀察到的自發鈣瞬變趨勢類似。與對照組相比,DMD hiPSC-CMs表現出鈣瞬變異常,除Con2/DMD2外瞬變幅度降低、到達峰值的時間延遲、TD50延長,但衰減τ沒有差異,與先前在DMD和其他遺傳性擴張型心肌病hiPSC-CMs中表征的鈣瞬變類似。總之,使用兩種不同的鈣瞬變成像方法證實了DMD hiPSC-CMs鈣處理異常。
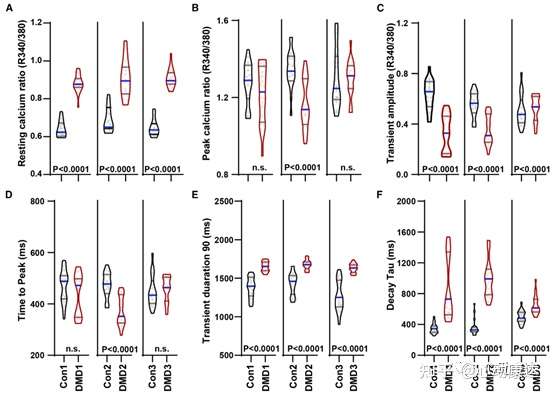
圖2 DMD hiPSC-CMs表現出鈣處理異常。(A)靜息,(B)峰值,(C)瞬移幅度,(D)到達峰值的時間,(E)90%鈣瞬變持續時間,(F)衰減τ。
02-當受到增加的機械負荷時,DMD hiPSC-CMs表現出收縮力產生障礙
為測量DMD hiPSC-CMs的收縮力,開發了一個牽引力顯微鏡平臺,使用可調水凝膠模擬纖維化發生前(10kPa)和后(35kPa)的心臟組織硬度(圖3A)。通過微圖案化將單個hiPSC-CMs構建為7:1長寬比,這是成體心肌細胞的特征,可促進HiPSC-CMs肌節排列和成熟。牽引力顯微鏡平臺捕捉單個心肌細胞產生的力,作為水凝膠變形的函數,通過跟蹤嵌入的熒光微珠(圖3B),使用傅立葉變換牽引細胞術重建牽引應力。測量DMD和10和35kPa水凝膠裝置上培養的健康hiPSC-CMs中,幾個收縮周期中的收縮參數,包括力和速度(圖3C。
35kPa條件下,與對照組相比,DMD hiPSC-CMs中力的產生顯著減少,但10kPa水凝膠中沒有(圖3D)。35kPa水凝膠上的DMD hiPSC-CMs還顯示收縮速度(圖3E)、舒張速度(圖3F)和細胞表面積(圖3G)降低,與鈣處理方面的測量結果一致。僅在35kPa基質上培養的DMD hiPSC-CMs中出現收縮缺陷,這一事實表明一旦心臟組織經歷纖維化硬化(DMD心肌病的一個標志),DMD心肌細胞就無法補償dystrophin缺陷。
實驗測得基礎條件下,DMD hiPSC-CMs的活性氧(ROS)增加,但線粒體呼吸沒有差異。已知心肌細胞在心力衰竭條件下會轉變為糖酵解代謝。實驗發現在營養缺乏條件下,如僅含葡萄糖、僅含丙酮酸鹽或僅含棕櫚酸鹽,DMD hiPSC-CMs基礎耗氧率和最大耗氧率顯著降低。表明DMD hiPSC-CMs表現出代謝適應不良和收縮功能障礙,與DMD心力衰竭進展中的觀察結果類似。
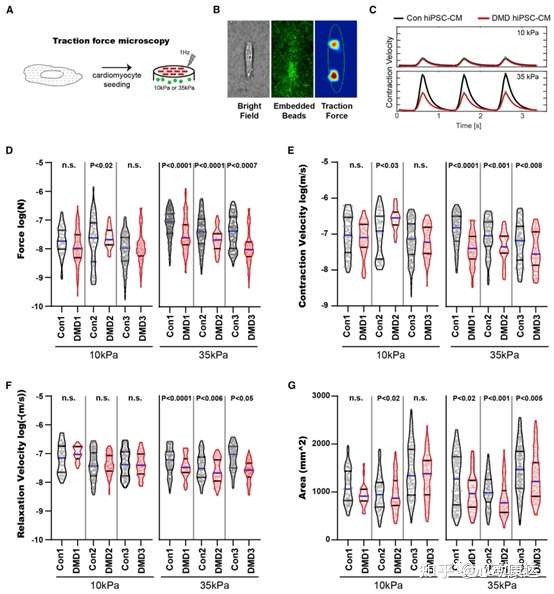
圖3 纖維化微環境處理下DMD hiPSC-CMs表現出收縮功能障礙。(A)牽引力顯微鏡評估DMD hiPSC-CMs收縮。(B)10或35kPa水凝膠上單個hiPSC-CMs產生的收縮力。(C)收縮周期。(D)力,(E)收縮速度,(F)舒張速度和(G)細胞面積。
03-沒有細胞分裂情況下的端粒縮短
探究了DMD hiPSC-CMs是否會表現出端粒縮短,與人DMD心臟組織心肌細胞和mdx4cv/mTRG2心臟組織類似。使用Q-FISH測量了一段時間內增殖性hiPSCs和分化的hiPSC-CMs的端粒信號(圖4A和4B)。增值性hiPSCs的端粒信號與健康對照和DMD hiPSCs,或三個等基因對間無統計學差異(圖4C)。
伴隨著衰老過程中每個細胞分裂的不完全復制,端粒損失通常是一個“被動”過程。通過hiPSC-CM平臺能夠測試心肌細胞在沒有DNA復制和細胞分裂的情況下是否也會發生端粒縮短。已知小鼠和人出生后發育期間,心肌細胞大多處于有絲分裂后期。通過EdU摻入法測定端粒長度及DNA復制,評估了hiPSC-CMs分化的一段時間,其中第0天被定義為分化的第一天(圖4D、4E)。發現與DMD小鼠(mdx4cv/mTRG2)和DMD患者結果一致,DMD hiPSC-CMs端粒較對照組逐漸縮短,在第30天端粒減少了50%(圖4D)。EdU標簽顯示,在第16天仍發生增殖,但在第20天至第30天之間增殖停止(圖4E)。正是在有絲分裂后的這段時間里,觀察到DMD hiPSC-CMs端粒與對照組相比發生進行性減少,最終在第30天減少了約50%(圖4D)。
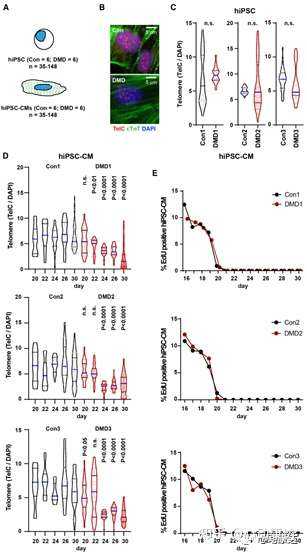
圖4 DMD hiPSC-CMs表現出端粒縮短和DNA損傷應答。(A)Q-FISH量化端粒長度(TelC)。(B)心臟troponin T、TelC、DAPI染色的hiPSC-CMs。(C)Q-FISH量化hiPSC端粒。(D)第20-30天間DMD hiPSC-CMs逐漸出現端粒丟失。(E)第20-30天hiPSC-CMs缺乏EdU。
假設端粒縮短的早期分子觸發因素可能是端粒失去覆蓋的外殼蛋白。研究人員分離了第20天,即非增殖出現端粒縮短時的心肌細胞,TMRM染色評估線粒體膜電位,流式細胞儀富集hiPSC-CMs。qRT-PCR顯示與對照hiPSC-CMs相比,端粒縮短DMD中端粒重復結合蛋白(TRF1和TRF2)和shelterin復合蛋白(RAP1、TIN2和POT1)的轉錄水平降低。為確定端粒縮短是否激活了DNA損傷應答,評估了p53水平(圖5A)、每個單細胞p53結合蛋白1(53BP1)foci數(圖5B和5D)和p21水平(圖5C和5E)。發現DMD hiPSC-CMs中p53蛋白上調(圖5A),對照組和DMD組中53BP1陽性hiPSCCMs的發生率從10%增加到50%(圖5)。此外,53BP1的積累與p53下游靶向p21蛋白的表達相關(圖5C和5E)。如53BP1升高所示,DNA損傷應答的激活誘導p53介導的PGC-1a抑制,這反過來阻礙線粒體生物發生(圖5F-5H)。另外與健康對照組相比,DMD hiPSC-CMs中凋亡標記物caspase-3和cleaved PARP增加,β-半乳糖苷酶信號積累增加。當從第20天開始每天使用布雷他汀(blebbistatin)阻斷DMD hiPSC-CM收縮,通過控制劑量將肌球蛋白頭部鎖定在低親和力狀態,從而防止actin結合,發現端粒縮短終止了,第30天測量確定確實如此(圖5I)。表明shelterin基因下調伴隨著由于異常收縮導致的非細胞分裂的端粒縮短,引起p53依賴的DNA損傷應答。
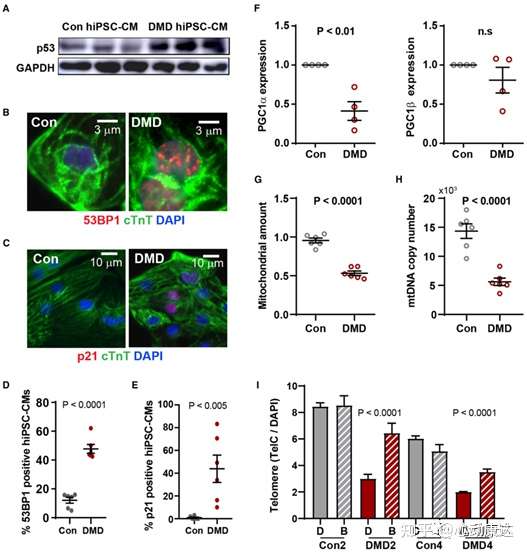
圖5 DMD hiPSC-CMs中p53上調。(A)p53激活的western圖像。(B)DNA損傷53BP1 foci的免疫熒光圖像。(C)心臟troponin T陽性hiPSC-CMs中的p21。(D, E)p21定量。(F)qRT-PCR測得PGC-1α(線粒體生物發生的主要調節因子)表達減少。(G)MitoTracker Green和qRT-PCR測得的線粒體數量。(H)線粒體拷貝數量。(I)第20-30天使用布雷他汀抑制DMD hiPSC-CMs收縮時,可阻止端粒丟失。
結論
本研究證明了在含結構蛋白dystrophin突變的hiPSC源心肌細胞中,端粒會通過一種復制非依賴性機制進行性縮短。已知DMD小鼠模型(mdx4cv)中端粒長度的人源化可以顯露出擴張型心肌病表型,在Noth單倍體不足(Notch+/-)小鼠中科顯露雙尖瓣表型。使用DMD hiPSC疾病模型能夠繪制端粒縮短的動態圖,證實并擴展了在鼠和人心臟組織中觀察到的致病過程。
文中的DMD hiPSC-CM模型和牽引力顯微鏡在硬水凝膠上的應用如實地概括了DMD患者心肌病的特征。不僅探究了DMD心肌纖維化發展后期,硬度增加損害DMD hiPSC-CMs收縮功能的機制,還表明心肌細胞可以補償健康心臟中dystrophin的缺乏,但無法補償僵硬、纖維化的心臟,這可以解釋為什么DMD患者隨著年齡的增長越來越容易患心律失常和心力衰竭。對不同硬度微環境的收縮反應的特征表明,DMD心肌細胞急性機械功能障礙的發展與心臟纖維化進展是同步的。
端粒縮短的分子觸發因素可能是shelterin復合物的丟失。研究發現DMD心肌細胞中,shelterin復合物與端粒的結合在非增殖性端粒縮短開始時減少,這可能是由于缺乏dystrophin時的收縮應激。鑒于DMD hiPSC-CMs中ROS產生增加約30%,且已知高ROS可以驅動端粒氧化并破壞shelterin結合,因此可推測未被覆蓋的端粒重復序列的直接氧化也可能參與端粒縮短。雖然研究表明shelterin在DMD hiPSC-CMs中的表達降低,但仍有待確定shelterin表達的喪失是DMD心肌細胞端粒去保護的原因還是結果。此外,ROS的產生部分由DMD心肌細胞收縮活動驅動。研究發現布雷他汀阻礙端粒縮短,表明收縮功能至關重要,但這是否是由于ROS介導的端粒去保護或肌節組織破壞仍有待闡明。盡管如此,這些數據仍支持收縮與關鍵結構收縮蛋白的遺傳缺失是端粒縮短的主要原因。另外DMD hiPSC-CMs中結構完整性的喪失可能會引發轉錄變化,下游后遺癥包括激活DNA損傷應答(53BP1、p53和p21蛋白積累)、線粒體死亡和代謝衰竭,與mTRG4小鼠衰老模型和DMD小鼠“人源化”端粒模型(mdx4cv/mTRG2)一致。
DMD hiPSC-CMs獲得“衰老樣狀態”是由于53BP1、p53、p21和β-半乳糖苷酶的表達。雖然端粒長度在心肌病發病機制中非常關鍵,但DNA損傷應答可能會破壞對細胞存活重要的其他途徑,如抑制線粒體生物發生相關的PGC-1α。
總之,研究表明營養不良的心肌細胞中,端粒縮短的發生與增殖無關,并且可能由收縮誘導的ROS驅動。這提出了一個治療靶點,端粒維持,對改善或阻止DMD心肌病進展有很大推進作用。
參考文獻:
Chang, Alex C. Y. , et al. "Increased tissue stiffness triggers contractile dysfunction and telomere shortening in dystrophic cardiomyocytes." (2021).



