
PAGA在monocle2擬時序分析中的應用


目前進行細胞軌跡分析的方法和軟件非常之多,常見的軌跡分析主要是通過monocle軟件為代表的擬時序分析(pseudotime analysis)。事實上monocle提供了一套具有啟發意義的軌跡方法,通過簡單粗暴的方式試圖彌補這理想與現實的間隙。在monocle軟件里軌跡與圖譜是分離的,即TSNE/UMAP的圖譜以及另一個降維空間的軌跡。那么有沒有一種降維技術能夠把兩者結合起來呢?今天我們介紹的scanpy的PAGA(graph abstraction reconciles clustering with trajectory inference through a topogy preserving map of single cells)寄希望在保留細胞圖譜的基礎上完成細胞軌跡的推斷,從而在聚類核軌跡上實現了統一。
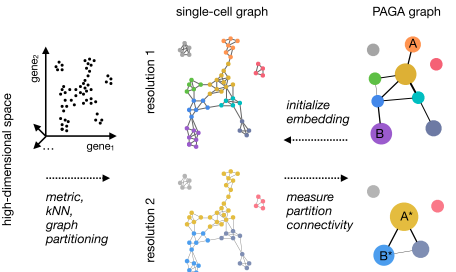
圖1 基于分區的圖抽象生成保留拓撲的單細胞映射
PAGA可以被看作是一種易于解釋和穩健的拓撲數據分析方法,通過高維基因表達數據降維后計算鄰域關系的相關距離度量來表示kNN圖。將kNN圖以期望的分辨率進行分區,其中分區表示連續的細胞群(partitions represent groups of connected cells)。為此,可以使用Louvain算法,當然也可以通過其他方式進行分區。接下來,PAGA圖通過將一個節點與每個分區關聯起來,并通過分區間連同性的統計度量的加權邊連接每個節點。然后,通過丟棄低權重的假邊,PAGA圖揭示了數據在選定分辨率下的去噪拓撲,并揭示了其連接和斷開的區域。是不是感覺很繞,簡單來說就是,點代表一個細胞類群,兩點之間的連線代表兩個細胞類群之間有關系,線的長短反映兩個細胞類群在聚類圖上的位置關系(請選擇忽視),線的粗細表示得到的軌跡關系的置信度(請選擇重視),線越粗,置信度越高。
接下來,我們一起來看看scanpy中PAGA的效果怎么樣吧。
最近,Plass等人利用PAGA對來自21,612個細胞的scRNA-seq數據重建了整個成年動物的細胞譜系。作者研究了使整體連通性最大化的樹狀子圖(通過逆PAGA連通性加權的G∗的最小生成樹),同時,作者還展示了如何將PAGA用于生成具有多種分辨率的數據映射圖。同流形學習(連接的組織類型以斷開或重疊的形式出現)相反,每個映射都保留了數據的拓撲。PAGA的多分辨率功能直接解決探索性數據分析,特別是對于單細胞數據:需要更高細節的區域重新聚類。

圖2 PAGA應用于整個扁蟲的圖譜
盡管PAGA圖中的連接通常對應于實際的生物軌跡,但情況并不總是如此。這是PAGA應用于kNN圖的結果,它只包含關于數據拓撲的信息。因此,我們不禁思考,既然每個生命從長遠來看所有的細胞都來自一個細胞,是不是在一套數據集中可以設置一個遙遠的點作為發育的起點呢?這樣是不是更能反映軌跡推斷的實際呢?
因此,有人考慮基于RNA速率的有向圖,用于存儲有關細胞轉變的信息。為了實現這一目的,我們將無向的PAGA連接性度量擴展到此類有向圖,并使用它來定向PAGA圖中的邊。由于高維特征空間scRNA-seq數據的特殊性,直接擬合RNA速率向量是很困難的。
PAGA是否能夠提供了一種可以提取拓撲信息和RNA速率信息的方法?
接下來,通過對Wagner等人的斑馬魚數據進行分析,將PAGA用來分析斑馬魚胚胎在不同發育時間點收集的53,181個細胞。PAGA圖準確地獲取了時間進展的鏈拓撲,并且更容易解釋的細胞類型譜系關系。通過對PAGA坐標初始化ForceAtlas2布局自動產生的單細胞數據。將精細細胞類型的PAGA圖與的粗線度圖進行比較,再現了作者之前的結果。
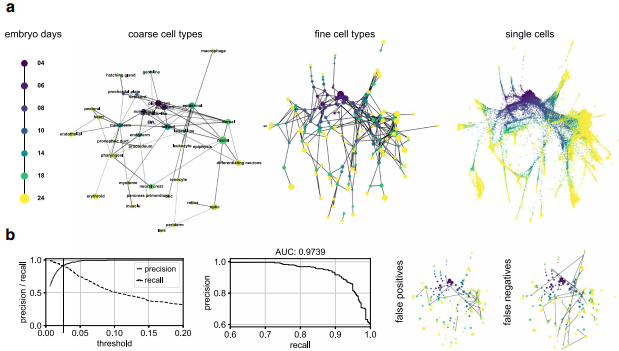
圖3 PAGA應用于斑馬魚胚胎數據
小編小結
scRNA-seq近些年的發展對生物醫學研究領域產生極大的影響。由此產生的數據集也是海量的,然而,對這些景觀(landscape)數據反映細胞異質性和模式的算法分析,仍然面臨著巨大的挑戰。目前的計算方法通常以如下兩種方式之一來解決這一點 :細胞聚類和細胞軌跡分析。雖然前者是大多數單細胞數據分析的基礎,而后者可以更好地解釋連續表型和過程。PAGA基于圖形的分析方法把這兩種分析思路進行了統一,先通過Louvain algorithm算法對細胞進行降維,生成低緯度的聚類圖,基于聚類圖進一步分析不同細胞類群之間的關系。所以,與其說PAGA是軌跡分析圖,不如說是軌跡關系圖。
參考文獻
F. Alexander Wolf et al,PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells.Genome Biol . 2019.doi:10.1186/s13059-019-1663-x.




