
ģųŌōÖίĶÖŦFĘīšâŖŦęPĶÚĩØØÄãÖĒļāÉŲ
12ÔÂ7ČÕ-10ČÕŖŦŌģÄęŌģļČĩÄÃĀøŅĒŌēWūŖ¨ASHŖŠČįÆÚļøÖÁŖŦĘĸūÉĪí×ÔČĢĘĀŊįĩÄŅĒŌēWš¤×÷Õß°ląíÖø×îĐÂßMÕšĄŖČį°ŨļúšĢ˛ŧÁËÆäAAVģųŌō¯ˇ¨ÔÚAĐÍŅĒĶŅ˛ĄĩÄ1/2ÆÚÅR´˛ÔōÖĐČĄĩÃˇeOĩūŖŦSangamošĢ˛ŧáĻβĩØØĩÄ1/2ÆÚÅR´˛×îĐÂĩūŖŦß@ĘĮĀ^CRISPR TherapeuticsēÍVertex PharmaceuticalsĐû˛ŧÆäCTX001ÔÚÅR´˛1/2ÆÚÔōĻŨŅĒŌĀŲĐÔβĩØÖĐēŖØŅĒ°YŖ¨TDTŖŠēÍĀÖØį îŧ°ûØŅĒ°YŖ¨SCDŖŠÖĐĩÄĶĐЧÖίĩūēķĩÄŖŦģųŌōÖίÔÚßz÷îŅĒŌēŧ˛˛ĄÖĐĶÖŌģŊŨķĄŖÍŦrŊņÄę6ÔÂŖŦBluebird biošĢËžŌ˛Đû˛ŧÆäģųŌō¯ˇ¨ZyntegloąģWÃËÅúĘÉĪĘĐÖίβĩØØĄŖÔŊíÔŊļāĩÄŋÆŅĐFę ŧĶČëģųŌōÖίĩØØĩÄŅĐ°lę ÎéŖŦŧČ×ÎŌŋ´ĩŊģųŌō¯ˇ¨ÖÎĶúßz÷ŧ˛˛ĄĩÄÄÜŖŦÍŦrŌ˛cĐŌß@îÕæÕũ“ČąátÉŲË”ĩÄēąŌŧ˛˛ĄÖđuĘÜĩŊÉįūVˇēęP×ĸĄŖËųŌÔŖŦąžÆÚÎŌžÍÅc´ķŧŌŌģÆđ×ßßMß@î˛ģąģ´ķąĘėĪ¤ĩÄŧ˛˛Ą—ĩØÖĐēŖØŅĒŖŦÖØücęP×ĸÆäÔÚøČĩÄĮérĄŖ
ŌģĄĸĘ˛Ã´ĘĮßz÷ĐÔŅĒŌē˛Ą
ßz÷ĐÔŅĒŌē˛ĄŖ¨hereditary blood diseasesŖŠĘĮÖ¸ßz÷ŌōËØŌũÆđĩÄŅĒŌēĪĩŊyÔėŅĒšĻÄÜÎÉyĩÄŧ˛˛ĄŖŦÖ÷ŌĒ°üĀ¨ßz÷ĐÔŧtŧ°ûĪĩŊyŧ˛˛ĄŖŦßz÷ĐÔ°×ŧ°ûĪĩŊyŧ˛˛ĄŌÔŧ°ßz÷ĐÔŗöŅĒŧ˛˛Ą×´ķîĄŖÆä´úąíĐÔŧ˛˛ĄÖ÷ŌĒĶĐĩØÖĐēŖØŅĒŖ¨ThalassemiaŖŦēˇQĩØØŖŠŖŦį ĩļĐÎŧtŧ°ûØŅĒ°YŖ¨Sickle Cell Disease, SCDŖŠŅĒĐĄ°åoÁĻ°YŖ¨Glanzmann thrombastheniaŖŠŖŦŅĒĶŅ˛ĄŖ¨HemophiliaŖŠĩČ[1]ĄŖÆäÖĐĩØØēÍSCDžųŲĶÚģųŌōÍģ×ĘšŅĒŧtĩ°°×ČąĪŨļø§ÖÂĩÄßz÷ĐÔŧtŧ°ûĪĩŊyŧ˛˛ĄŖŦšĘŗŖŋÉŌÔĶÃĶÚģųŌō¯ˇ¨¸ųÖÎŖĄČįCTX001˛ÉĶÃCRISPR/Cas9ŧŧĐgžŨ˛ĄČË×ÔķwCD34+ÔėŅĒ¸Éŧ°ûēķģØŨ˛ĄČËķwČŖŦÄļøÔ´Ô´˛ģāĩÄŽaÉúÕũŗŖŅĒŧtĩ°°×ŌÔß_ĩŊÖίÉõÖÁÖÎĶúĩØØēÍSCDĩÄЧšûĄŖ
ļūĄĸĩØØēÍį îŧ°ûØŅĒ°YŲĶÚēąŌ˛ĄáŖŋ
1. ĩØØēÍį îŧ°ûØŅĒ°YĩÄ°l˛ĄŦF î
ĩØØēÍį îŧ°ûØŅĒ°YŲĶÚēąŌ˛ĄĩÄÕfˇ¨ËÆēõŌŅŊąģVˇē÷˛ĨŖŦ˛ģß^ß@ˇNÕfˇ¨ßĶĐ´ũŋŧÁŋĄŖĪČŋ´ŌģŊMĩūŖē1925ÄęŖŦøëHÉĪCooleyēÍLee Ę×ĪČÃčĘöĩØÖĐēŖØŅĒŖģÎŌøĶÚ1940ÄęĘ×´ÎķĩĀVÖŨ3ÃûĩØØģŧÕßŖŦļøēķęĀm°lŦFĶÚąąžŠŖŦÕãŊĩČĩØ ^ŖŦ20ĘĀŧoÖĐÆÚŖŦŌģ´Î¸˛ÉwČĢø20ĘĄĘĐŖ¨×ÔÖÎ ^ŖŠĩÄ90ČfČË´ķŌÄŖŅĒŧtĩ°°×˛ĄÕ{˛éŖŦģųąžęUÃ÷ÁËÖĐøéLŊŌÔÄĪĘĮĩØظ߰l ^ŖŦĶČŌÔVÎ÷ŖŦV|ēÍēŖÄĪČũĘĄŖ¨ ^ŖŠéÉõ[2,3]ĄŖ
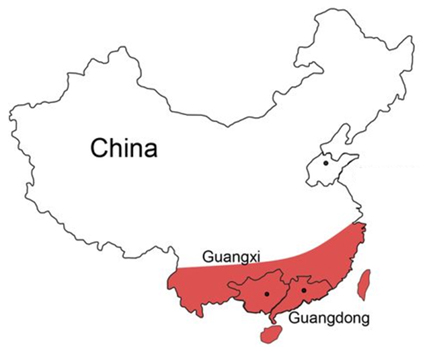
D1ŖēÎŌøĩØØÖ÷ŌĒˇÖ˛ŧ ^Ķō[2]
ūĄļÖĐøĩØÖĐēŖØŅĒˇĀÖÎË{ƤøŖ¨2015ŖŠĄˇĩÄĩūī@ĘžŖŦÄŋĮ°ÎŌø“ĩØØ”ģųŌōy§Õßŧs3000ČfČËŖŦÉæŧ°Ŋü3000ČfŧŌÍĨŌģ|ČËŋÚŖŦÆäÖĐÖØĐÍēÍÖĐégĐÍ“ĩØØ”ģŧÕßŧs30ČfČËĮŌÕũŌÔÃŋÄęŧs10ŖĨĩÄËŲļČßfÔö[3]ĄŖ
ĩØØÖ÷ŌĒˇÖ˛ŧÔÚ°üĀ¨ÖĐøÄĪˇŊÔÚČĩÄČĢĮō¯ŧ˛¸ß°lĩÄá§ēÍá§ĩØ ^ŖŦÎŌøéLŊŌÔÄĪV´ķĩØĶōĘĮĩØÖĐēŖØŅĒĩĸ߰l ^ŖŦĶČÆäŌÔVÎ÷ĄĸV|ēÍēŖÄĪČũĘĄéÉõĄŖÆäÖĐVÎ÷ĩØØ°l˛ĄÂĘ×î¸ßŖŦČËČēÖĐÃŋ4~5ČËžÍĶĐ1ĩØØČąĪŨģųŌōy§ÕßŖŦÃŋ55ŧŌÍĨžÍĶĐ1ĶĐÖØĐÍĩØØŗöÉúīLëUŖŦČįšû]ĶĐĀ¸ņĩġĀŋØ´ëĘŠŖŦÃŋŗöÉú200~250ĖĨēžÍĶĐ1ÖØĐÍĩØØģŧēŖģÆä´ÎĘĮV|ŖŦĩØØģųŌōy§Õß´ķĶÚ10%ŖŦŌÔ2019ÄęČËŋÚĩ1.13|ČËĶËãŖŦĩØØģųŌōy§ÕßŗŦß^1000ČfČË[3]ĄŖ
2. ČįēÎļ¨ÁxēąŌ˛ĄŖŋ
ģØĩŊé_ĘŧĩÄî}ÉĪŖŦęPĶÚēąŌ˛ĄĩÄļ¨ÁxŖŦĘĮÖ¸°l˛ĄÂĘēÜĩÍĄĸēÜÉŲŌĩÄŧ˛˛ĄŖŦŌģ°ãéÂũĐÔĄĸĀÖØĩÄŧ˛˛ĄĄŖĩĢ˛ģÍŦøŧŌļ¨Áx˛ģÍŦŖē

ąí1. ˛ģÍŦøŧŌĻĶÚēąŌ˛ĄĩÄļ¨Áx[4]
ĶÉÉĪąíŋ´ĩŊŖŦ˛ģÍŦøŧŌĻĶÚēąŌ˛Ąļ¨ÁxŌĀøĮéļø˛ģÍŦŖŦĩĢÔÚÖĐøŖŦēąŌ˛ĄŌģÖą]ĶĐÃ÷´_ĩġ¨ÂÉáÁxĄŖ2010Äę5 ÔÂŖŦÖĐČAátWūátWßz÷WˇÖūÔÚÉĪēŖÅeŪkĩÄÖĐøēąŌ˛Ąļ¨ÁxŖŧŌŅĐĶūÉĪŖŦÅcūŖŧŌŊ¨×hĸÖĐøĩÄēąŌ˛Ąļ¨Áxéģŧ˛ĄÂĘĐĄĶÚ1/500,000ģōĐÂÉúē°l˛ĄÂĘĐĄĶÚ1/10,000 ĩÄŧ˛˛ĄĄŖū´ËšĀËãŖŦÖĐøēąŌ˛ĄģŧÕßČËĩŧsé1,680ČfĄŖČįšû°´ÕÕ30ČfĩÄ°l˛ĄČËĩĶËãŖŦĩØØēôķĶÚß@°l˛ĄÂĘĄŖ
3. ĩØØŋÉÄܲĸ˛ģŲĶÚēąŌ˛ĄŖŦļøį îŧ°ûØŅĒ°Y sÔÚÁĐŖĄ
2018Äę5ÔÂŖŦøŧŌĐlÉúŊĄŋĩίTūĄĸŋÆWŧŧĐg˛ŋĄĸš¤IēÍĐÅĪĸģ¯˛ŋĄĸøŧŌËÆˇąOļŊšÜĀížÖĄĸøŧŌÖĐátËšÜĀížÖÎå˛ŋéTÂēĪšĢ˛ŧĄļĩÚŌģÅúēąŌ˛ĄÄŋ䥡ŖŦĘÕäÁË121ˇNēąŌ˛ĄĄŖß@ĘĮÖĐøÕū¸ŽĘ×´ÎŌÔŧ˛˛ĄÄŋäĩÄĐÎĘŊŊįļ¨ēÎéēąŌ˛ĄĄŖÔÚß@ÁĐąíĀīŖŦÎŌ˛ĸ]ĶĐŋ´ĩŊĩØØŖŦ˛ģß^į îŧ°ûØŅĒ°Y sÔÚÁĐĄŖŌōéÔÚÎŌøŖŦį îŧ°ûØŅĒ°Y°l˛ĄÂĘēÜĩÍŖŦÖ÷ŌĒŧ¯ÖĐÔÚēÚÉĢČˡNŖŦČįÔÚˇĮÖŪēÚČËÖĐĩÄ°l˛ĄÂĘ×î¸ßŖŦÔÚŌâ´ķĀûŖŦĪŖÅDĩČĩØÖĐēŖŅØ°ļøŧŌēÍĶĄļČĩČĩØŖŦ°l˛ĄČËĩŌ˛˛ģÉŲŖŦÄŋĮ°Ö÷ŌĒÔÚÎŌøÄĪˇŊ°lŦFß@î°¸ĀũĄŖËųŌÔĩØØĘĮˇņŲĶÚēąŌ˛ĄŌģÖą]ĶĐÃ÷´_Õfˇ¨ĄŖÖÁÉŲÄŋĮ°ĩūŋ´íŖŦĩØØ°ČģŌŅŗÉéÎ÷ÄĪĩØ ^ĩÄŗŖŌ˛ĄŖŦŌō´ËĀ¸ņˇĀŋØŌÔŧ°Öί´ëĘŠĩÄÖØŌĒĐÔ˛ģŅÔļøÃ÷[5]ŖĄ
ČũĄĸĩØØĩÄÖ²ĄCĀí
ĩØØ×îÔį°lŦFĶÚĩØÖĐēŖČËČēŖŦšĘˇQéĩØÖĐēŖØŅĒŖŦĶÖˇQēŖŅķĐÔØŅĒģōÖéĩ°°×ÉúŗÉÕĪĩKĐÔØŅĒŖŦĶÉĶÚÖéĩ°°×ģųŌōąíß_ŽŗŖļøoˇ¨ĐÎŗÉÕũŗŖšĻÄÜĩÄŅĒŧtĩ°°×ļøŌũ°lŖŦĘĮŌģˇNŗŖČžÉĢķwęĐÔßz÷˛ĄŖŦĘĮ×îÔįÔÚˇÖ×ĶËŽÆŊÉĪęUĘöÆ䲥ĀíWCÖÆĩÄČËîßz÷˛ĄÖŽŌģĄŖžßķwÖ²ĄCĀí[6]ČįĪÂŖē
ČįĪÂDËųĘžŖŦČËķwÕũŗŖŅĒŧtĩ°°×ŊYĶĐ2lα-Öéĩ°°×ēÍ2lβ-Öéĩ°°×ˇÖey§ŌģŅĒŧtËØēķžÛēĪĐÎŗÉŅĒŧtĩ°°×Ŗ¨hemoglobinŖŦHbŖŠËÄžÛķwŖŦŗĐúČËķwČŅõß\ŨČÎÕŖŦŽα-Öéĩ°°×Åcβ-Öéĩ°°×ąČĀũŖ¨ÕũŗŖąČĀũéαŖēβ=1ŖŠĘ§ēâŖŦĩØØ°l˛ĄĄŖ
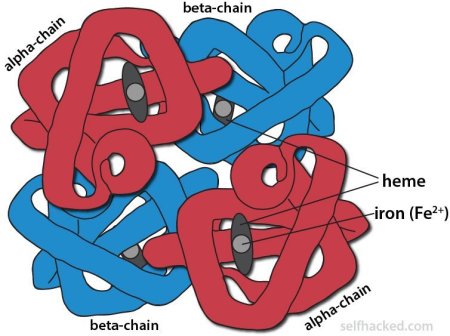
D2. ČËîÕũŗŖÖéĩ°°×ģųŌōĘžŌâD
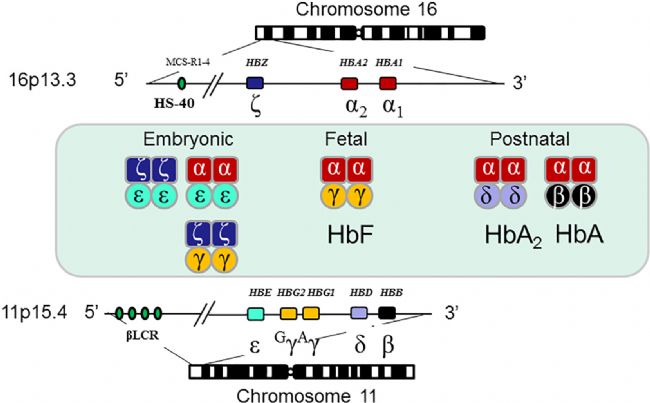
D3. ČËîα-ēÍβ-Öéĩ°°×ģųŌōļ¨ÎģÅcŊY[6]
Ąž×ĸĄŋŖēα-Öéĩ°°×ģųŌōÎģĶÚ16ĖČžÉĢķw16p13.3ÎģücŖŦąžģųŌō´ØēŦĶĐÅßĖĨÆÚąíß_ģųŌōŖ¨ζŖŠŌģŖŦĖĨēÅcŗÉČËÆÚąíß_ģųŌōŖ¨α1ēÍα2ŖŠ2ŖŦŧŲģųŌōŖ¨ψζŖŦψα1ŖŠ2ŖŦŌÉËÆÖéĩ°°×ŧŲģųŌōŖ¨ψα2ŖŦθŖŠ2ŖŦŌÔÉĪα×åģųŌōĩÄąíß_ĘÜŋØĶÚÖØŌĒÕ{ŋØÎģücHS-40ĄŖÍŦĀíβ-Öéĩ°°×ģųŌō´ØÎģĶÚ11ĖČžÉĢķw11p15.3ÎģücŖŦÅÅÁĐĶĐÅßĖĨÆÚąíß_ģųŌōŖ¨εŖŠ1ŖŦĖĨēÆÚąíß_ģųŌōŖ¨ GγŖŦαγŖŠ2ŖŦŗÉČËÆÚąíß_ģųŌōŖ¨βēÍδŖŠ2ŖŦŧŲģųŌōŖ¨ψβŖŠ1ŖŦŌÔÉĪβ×åģųŌōąíß_ĘÜŋØĶÚ×ųÎģÕ{ŋØ ^Ŗ¨LCR, locus control regionŖŠ[6]ĄŖ
1. ĩØØĩġNî
¸ųūŽŗŖąíß_ģųŌōĩIJģÍŦŖŦŋɡÖéα-ĩØØŖŦβ-ĩØØŖŦδ-ĩØØŖŦγ-ĩØØŖŦδγ-ĩØØŖŦεγδβ-ĩØØŖŦÆäÖĐα-ēÍβ-ĩØØŨ^éŗŖŌĄŖ´ËÍâŌĀū˛ĄĮéŨpÖØ˛ģÍŦŖŦβ-ĩØØĶÖˇÖéÖØĐÍŖŦÖĐĐÍēÍŨpĐÍŖŦα-ĩØØĶÖˇÖéėoÖšĐÍŖŦŨpĐÍŖŦÖĐégĐÍēÍÖØĐÍĄŖžßķwČįĪÂąíËųĘž
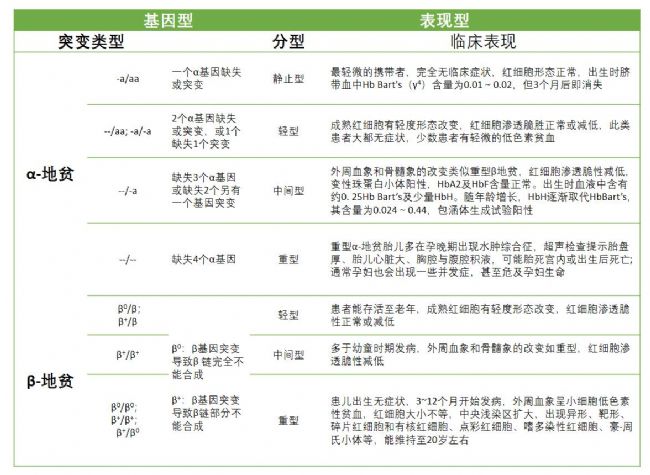
ąí2. ¸ųūģųŌōĐÍĻĩØØßMĐСÖîŧ°ÆäąíŦFĐÍ[6]
2. ĩØØĩÄÖ÷ŌĒÍģ×ĐÎĘŊ
α-ēÍβ-ĩØØŨ^éŗŖŌŖŦĪÂÃæÖ÷ŌĒŋ´ß@2ˇNĩØØĩÄÍģ×ĐÎĘŊĄŖα-ĩØØŖēÖ÷ŌĒĶÉģųŌōȹʧŌũÆđĩÄŖŦČįĪÂąíŖ¨-α3.7/ŖŠēÍŖ¨-α4.2/ŖŠĘĮÖĐøČË×îŗŖŌĩÄα+-ĩØØŖ¨α-ģųŌōšĻÄܲŋˇÖąŖÁôĶéα+ŖŦšĻÄÜÍęČĢĘʧĶéα0ŖŠŖģŗũÁËģųŌōȹʧŖŦŌ˛ĶĐÉŲ˛ŋˇÖĘĮĶÉģųŌōücÍģ×ŌũÆđĩÄŖŦÄŋĮ°ÔÚÖĐøŌŅķĩĀÁË12ˇNˇĮȹʧĐÍα-ĩØØŖŦÆäÖĐαWSα/Ŗ¨Hb WestmeadŖŦėoÖšĐÍα-ĩØØŖŠ×îéŗŖŌĄŖ
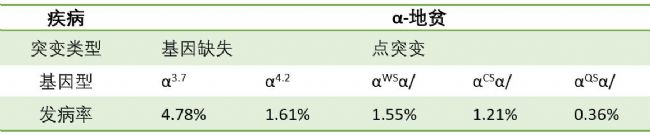
ąí3. α-ĩØØÖ÷ŌĒ˛ģÍŦÍģ×ĐÎĘŊĩÄÕŧąČ
β-ĩØØŖēÖ÷ŌĒĶÉģųŌōücÍģ×ŌũÆđŖŦČĢĘĀŊį°lŦF200ļāˇNŖŦÖĐøŌŅķĩĀ48ˇNĄŖÆäÖĐŗŖŌÍģ×ĶĐ6ˇNŖŦCD41-42(-TCTTȹʧ)ŖŦŧsÕŧ45ŖĨŖŦÆä´ÎĘĮIVS-II654ŖŦCD17(AĩŊTücÍģ×)ŖŦTATAēĐ×Ķ-28ĩČĄŖČįĪÂąíËųĘž
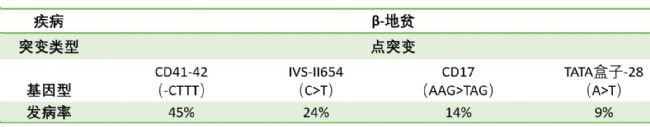
ąí4. β-ĩØØÖ÷ŌĒ˛ģÍŦÍģ×ĐÎĘŊĩÄÕŧąČ[6]
ËÄĄĸĩØØÖίĶСŊŖŋ
ÄŋĮ°ÅR´˛ÉĪáĻĩØØĩÄĖÖÃˇŊˇ¨ĶĐŨŅĒŖŦčFōüēĪŠÖίŖŦÆĸÅKĮĐŗũĩČĩČ[6]Ŗ¨éĘ˛Ã´ĶÓĖÖÔļøˇĮ“Öί”ŖŋÄĮĘĮŌōéŌÔÉĪˇŊˇ¨HHĘĮÆđĩŊžŊâÅcžSŗÖ×÷ĶÃĮŌ´æÔÚĀÖظą×÷ĶÃģōžÖĪŪĐÔŖŦ˛ĸ˛ģÄÜÖÎĶúŖŠĄŖ
- ŨŅĒŖēŨŅĒ¯ˇ¨éÄŋĮ°ĩØØÖ÷ŌĒĖÖôëĘŠŖŦÄŋĩÄÔÚĶÚžSŗÖÕũŗŖŅĒŧtĩ°°×ËŽÆŊŖŦîAˇĀÂũĐÔšŠŅõ˛ģ×ãĩČŌÔŧ°´úĐÔšĮËčÔöÉúĩȲĸ°l°YŖŦ´ËÍâéÁËąÜÃâŨŅĒˇ´ĒŖŦŨČëĩÄĘĮĪ´ėŧtŧ°ûŖŦŧ´ÉŲ°×ŧ°ûĩÄŧtŧ°ûŖŦĩĢˇ´ÍŨŅĒĘšķwČčFØēÉÔöŧĶŖŦ´æÔÚĀÖظą×÷ĶÃŖŦŧ´ēŦčFŅĒüSËØŗÁÖø°YŖŦąØíÍŦrŊĶĘÜČĨčFÖίŖģ
- ŗũčFÖίŖēČįÉĪËųĘöŖŦCķwčFØēÉÔöŧĶŖŦŧtĪĩŧ°ûÔėŅĒß^ĘŖŖŦÄcĩĀčFÎüĘÕŌ˛ūÔöŧĶŖŦCķwÅÅčFÕĪĩKŖŦČô˛ģŧ°rČĨčFŖŦūŌũÆđ°lĶũÍŖūŖŦ¸ÎĶ˛ģ¯ŖŦĖĮÄō˛ĄŖŦĐÄšĻÄÜËĨŊßĩČŖŦĩĢČĨčFÖίÍŦĶ´æÔÚĀÖØ˛ģÁŧˇ´ĒĄŖŗŖĶÃčFōüēĪŠ°üĀ¨ĩÃËšˇŌŖ¨DesferrioxamineŖŦČĨčF°ˇŧ×ģĮËáû}×ĸÉäŠŖģ˛ģÁŧˇ´ĒĶĐÖ°×ČÕĪŖŦéLšĮ°lĶũÕĪĩKĩČŖŠŖŦWØ°˛ŋÉŖ¨DeferiproneŖŦL1ŖŦČĨčFÍĒÆŦŖģ˛ģÁŧˇ´ĒĶĐęPšÍ´ŖŦÄcĩĀˇ´ĒēÍä\ČąˇĻĩČŖŠŖŦļ÷Čđ¸ņŖ¨DeferasiroxŖŦExjadeŖŦĩØĀÁ_ËžˇÖÉĸÆŦŖģ˛ģÁŧˇ´ĒĶĐθÄcĩĀˇ´ĒŖŦƤÕîŖŦ ĶXpÍËĩČŖŠŖģ
- ÆĸÅKĮĐŗũŖēĻĶÚŊŨŅĒŧ°ŌˇļĩÄČĨčFÖίļøčFØēÉČÔČģÔöŧĶĩÄģŧēŋŧ]ÆĸÅKĮĐŗũŖŦĩĢÆĸÅKĮĐŗũūÔöŧĶĀÖØĄŅĒ°Y°lÉúĩÄīLëUŖŦĮŌÆĸĮĐŗũĻÖØĐÍβ-ĩØØÖÎ¯Đ§šûČÔČģ˛îŖģ
-
ÔėŅĒ¸Éŧ°ûŌÆÖ˛Ŗ¨hematopoietic stemcell transplantation, HSCTŖŠŖē´Ëˇ¨ĘĮÄŋĮ°Î¨ŌģÄܸųÖÎÖØĐÍβ-ĩØØĩġŊˇ¨ĄŖŌÔČËî°×ŧ°ûŋšÔŖ¨HLAŖŠÅäĐÍßxņšŠķwŖŦˇŊĘŊĶĐšĮËčŌÆÖ˛ŖŦÍâÖÜŅĒ¸Éŧ°ûŌÆÖ˛ŖŦÄŅĒŌÆÖ˛ŖŦĩĢŌōēĪßmĩÄšŠķwíÔ´ĶĐĪŪĮŌr¸ņ°ēŲFÆŊžųát¯ŲMĶÃ40ČfÔĒ×ķĶŌŖŦÅR´˛ÉĪëyŌÔé_ÕšŖŦĮŌĶ°íŗÉšĻÂĘĩÄŌōËØˇąļāŖŦ˛ģHÅcÅäĐÍĶĐęPŖŦßĐčģŧÕß îBŧ´ŌŅŊĶĘÜĩÄÖίĶĐęPĄŖ´ËÍâŌÆÖ˛˛ĸ°l°YČįŌÆÖ˛ÎīŋšËŪÖ÷˛ĄŖ¨GVHDŖŠŖŦ¸ÎėoÃ}×čČû˛ĄŖ¨hepatic vein occlusive diseaseŖŦHVODŖŦēˇQVODŖŠĄĸ¸ĐČžĄĸŗöŅĒēÍŌÆֲʧĄžųÔöŧĶÁ˴ˡŊˇ¨ĩÄÆÕŧ°ëyļČĄŖ
ģųŌōÖί--ĩØØģŧÕßĩÄĪŖÍûŖĄ
ŊüČÕŖŦģųŌō¯ˇ¨ÔÚßz÷îŅĒŌēŧ˛˛ĄÖĐîl÷ŊŨķŖŦ×ÎŌŋ´ĩŊÁËģųŌōÖίÖÎĶúĩØØĩÄĪŖÍûĄŖÄŋĮ°ŖŦáĻĩØÖĐēŖØŅĒĩÄģųŌōÖί˛ßÂÔÖ÷ŌĒˇÖéɡNŖŦģųĶÚÂũ˛ĄļžÕûēĪĩġŊĘŊēÍģųĶÚģųŌōžŨĩġŊĘŊĄŖ˛ßÂÔéŖēßfËÍšĻÄÜĐÔβ-ŅĒŧtĩ°°×ģųŌōŋŊØģōģųŌōžŨŊMˇÖÖÁģŧÕßĩÄÔėŅĒ¸Éŧ°û(HSC)ŖŦÄ´Ë´úĖæģōŨoÖúŧmÕũ˛Ą×ĩÄβ-ŅĒŧtĩ°°×ģųŌōÍęŗÉš¤×÷ĄŖ×îĐÂßMÕš[7]ČįĪÂŖēCRISPR TherapeuticsēÍVertex PharmaceuticalsŖēÆäŲYÖúĩÄžĖéNCT03655678ĩÄA Safety and Efficacy Study Evaluating CTX001 in Subjects With Transfusion-Dependent ThalassemiaíÄŋŖŦÔÚÕũÔÚßMĐĐĩÄ1/2ÆÚÅR´˛ÔōÖĐČĄĩÃˇeOÖĐÆÚĩū, ŌģÃûŨŅĒŌĀŲĐÔβ-ĩØØģŧÕßēÍŌģÃûĀÖØį îŧ°ûØŅĒ°YģŧÕßÔÚŊĶĘÜCTX001ÖίēķŖŦžųß_ĩŊÍŖÖšŌĀŲŨŅĒĩÄЧšûĄŖ
ß@ĘĮÔÚÃĀøßMĐĐĩÄĘ×ÔušĀCRISPRģųŌōžŨ¯ˇ¨ĩÄČËķwÅR´˛Ôō;EditasšĢËžÉíÅR´˛Į°ōíÄŋŖēĶÚ2019Äę10ÔÂ7ČÕ°l˛ŧĪûĪĸŖŦÅcCas9ĻąČ¸üŧĶģ¯ĩÄCas12a-RNPŊ駞ŨCD34+×Ôķw¸Éŧ°ûģųŌōžŨĢ@ĩÃŋÉĪ˛ßMÕšŖŦĶĐÍûĶÃĶÚSCDÖίĄŖ´ËÍâĶÚ16Äę°l˛ŧCRISPR/Cas9ĶÃĶÚCD34+×Ôķw¸Éŧ°ûģųŌōžŨÖίĩØÖĐēŖØŅĒ;MemorialSloan Kettering Cancer CenterŲYÖúĩÄžĖéNCT01639690ĩÄß-ThalassemiaMajor With Autologous CD34+ Hematopoietic Progenitor CellsTransduced With TNS9.3.55 a Lentiviral Vector Encoding the Normal Humanß-Globin GeneíÄŋŖŦģųĶÚÂũ˛ĄļžŨdķwTNS9.3.55ÔŲžŨCD34+ŧ°ûŖŦÄŋĮ°ŌŅßMČë1ÆÚÔōëAļÎ;
Sangamo TherapeuticsŲYÖúĩÄžĖéNCT03432364ĩÄAStudy to Assess the Safety, Tolerability, and Efficacy of ST-400 for Treatment of Transfusion-Dependent Beta-thalassemia (TDT)íÄŋŖŦģųĶÚä\Ö¸ēËËáøŖ¨ZFNŖŠŧŧĐgŖŦÔŲžŨ˛ĄČË×ÔÉíŅĒŌē¸Éŧ°ûļøēķÔŲģØŨÄŋĮ°ŌŅßMČë2ÆÚÅR´˛ÔōĄŖÔÚŊüČÕĩÄASHÉĪŌ˛šĢ˛ŧÁË×îĐÂĩūŖŦąíÃ÷ģųŌō¯ˇ¨ÖĩĩÃßMŌģ˛ŊĖŊË÷;Bluebird bioŲYÖúĩÄžĖéNCT01745120ĩÄA Study Evaluating the Safety and Efficacy of the LentiGlobin BB305 Drug Product in β-Thalassemia Major ParticipantsíÄŋŖŦģųĶÚLentiGlobin BB305 lentiviral vectoržŨ˛ĄČË×ÔÉíCD34+ÔėŅĒ¸Éŧ°û ŌŅŊßMČë3ÆÚÅR´˛ŖŦĮŌÔÚŊņÄę6ÔÂŖŦģųŌōÖίËÎīZynteglo (LentiGlobin)ÔÚWÖŪĢ@ĩÃÅúĘÉĪĘĐŖ¨r¸ņé177ČfÃĀÔĒŖŠŖŦŗÉéĀ^5Ô¡Ũ FDAÅúĘÖίŧšËčĐÔŧĄÎŽŋsĩÄZolgensmaŖ¨é_°lÉĖéÖZČAŖŦr¸ņŧsé210ČfÃĀÔĒŖŠÖŽēķĩÚļūŋîŗŦß^100ČfÃĀÔĒr¸ņÉĪĘĐĩÄËÎīŖŦŌ˛ĘĮÄŋĮ°ČĢĮōĩÚļū°ēŲFĩÄËÎīŖģ
GlaxoSmithKlineŲYÖúĩÄžĖéNCT03275051ĩÄLong-term Follow-up of Subjects Treated With OTL-300 for Transfusion Dependent Beta-thalassemia Study (TIGET-BTHAL)íÄŋŖŦģųĶÚĶÞ´aČËβÖéĩ°°×ģųŌōĩÄÂũ˛ĄļžŨdķw(globe)ßz÷ĐŪīĩÄ×ÔķwÔėŅĒ¸Éŧ°û/×æŧ°ûˇÖģ¯´Ø(CD)34+ŧ°ûģØŨ˛ĄČËķwČŖŦÄŋĮ°ĖĶÚÅR´˛1/2ÆÚÔōëAļÎŖģIRCCS San RaffaeleŲYÖúĩÄžĖéNCT02453477ĩÄGene Therapy for Transfusion Dependent Beta-thalassemiaíÄŋŖŦģųĶÚGLOBE lentiviral žŨ˛ĄČË×ÔķwÔėŅĒ¸Éŧ°û˛ĸģØŨŖŦÄŋĮ°ĖĶÚÅR´˛1/2ÆÚÔōëAļÎĄŖęPĶÚĩØØĩÄŊéŊBąžÆÚžÍĩŊß@ĀīÁËŖŦ´ķŧŌĘĮˇņĻß@ŧ˛˛ĄĶÖĶĐÁËÖØĐÂĩÄÕJ×RÄØĄŖÃæĻģŧÕßČēķw˛ģāÉĪÉũŖŦĪŖÍûÎŌŋÉŌÔ˛ģāÍÆĶĩØØŧ°ÆäËûēąŌ˛ĄÔÚÉįūÉĪĩÄęP×ĸļČŖŦ׸üļāģŧÕßĢ@ĩÃÖίŖŦÖØˇĩÕũŗŖÉúģîĄŖ
ĸŋŧÎÄĢI
Ąž1ĄŋĀîģÛæÂ,îČĘŗØ.ßz÷ĐÔŅĒŌē˛ĄŅĐžŋßMÕš[J].ÖĐøŋÆW:ÉúÃüŋÆ W,2017,47(12):1306-1312.
Ąž2ĄŋČ~Īōģ¯ŖŦÎŌøĩÄĩØÖĐēŖØŅĒ°Y[J].čFĩĀátWŖŦ1980Ŗ¨09ŖŠ.
Ąž3ĄŋÖĐøĩØÖĐēŖË{ƤøŖ¨2015ŖŠ
Ąž4ĄŋÖĐøēąŌ˛ĄËÎīŋÉŧ°ĐÔķ¸æ,2019
Ąž5ĄŋÖĐøĩÚŌģÅúēąŌ˛ĄÄŋä,2018
Ąž6ĄŋĄļĩØÖĐēŖØŅĒîAˇĀŋØÖÆ˛Ų×÷Ö¸ÄĪĄˇŖŦÖ÷žŖēĐėĪæÃņ

- H5N1ĮŨÁ÷¸Đ˛Ąļž¸ĐČžīLëUÅcÔįÆÚzyˇŊˇ¨
- CDR3š¤ŗĖÅcÖĐēÍŋšķwļ¨ÖÆŖēąŖ×CĐÍŋšķwÖØËÜÖίĐÂËĘ
- ČįēÎŊ¨¸ßЧÍģ×øÎÄėŖŋ
- ŋšķwļ¨ÖÆĄĸŧ{Ã×ŋšķwÅcÅäĻŋšķwĩÄĐÂÁĻÁŋ
- ĘÉžúķwÕšĘžŧŧĐgŖēé_ĸÉúÎīátËĐÂŧoÔĒĩÄčŗ×
- Čũ´ķŧŧĐg fÍŦŖŦŊâæiŋšķwŅĐžŋĐÂδí
- ļāëÄÎÄėŊ¨ÅcēYßxˇūÕŖēŧĶËŲËÎīŅĐ°lĩÄēËĐÄōĶÁĻ
- ĘÉžúķwŊ¨ėÅcŋšķwĘÉžúķwÎÄėĩÄ fÍŦĐÂ
- ŅĐĶūŖēAIōĶŲ|×VŧŧĐgŲxÄÜÉúÎīËŅĐ°lÅcÃâŌßÖί
- °ŲWŲDŅûÄúĪāŧsChinaBioēĪ×÷Õ¯
- °ŲWŲDÅcMyricxß_ŗÉŋšķwÔušĀ f×hĶÚADCËÎīé_°l
- 2023Äę°ŲŨčÉúÎīŋÆŧŧÕ\ƸļāÎģ
- ×ŖŲR°ŲŨčÉúÎīÉĪēŖŅĐ°lÖĐĐÄĖßwĐÂÖˇÅcĐšŲžWÉĪž
- úˇ˛ÉúÎīÁÁĪāIND¸ĐČžĐÔŧ˛˛ĄĪČßMˇÖ×ĶÔ\āĒĶÃÕ¯
- ĩvēÍátËŋšķwÔÚČĢĮōIÆÚÅR´˛ŅĐžŋÍęŗÉĘ×ĀũģŧÕßŊoË
- °ŲWŲDÅcÄĪžŠÕũ´ķĖėĮįēĘđČĢČËŋšķwēĪ×÷é_°l f×h


