
質譜法進行高通量蛋白鑒定的主要方法介紹
蛋白質組(Proteome)指由一個基因組或一個細胞、組織表達的所有蛋白質。蛋白質全譜分析目的在于鑒定樣本中盡可能多的肽和蛋白質混合物的組分。基于質譜技術的全譜鑒定,可為蛋白高通量的定量和修飾分析提供參考信息。傳統的方法如蛋白質微量測序、氨基酸組成分析(如Edman降解法)費時費力、通量低,存在不容易實現規模化和自動化,結果靈敏度差等問題。當前主流的基于軟電離技術的液相色譜-質譜系統(LC-MS/MS)是實現高通量蛋白鑒定的主要方法。
其基本原理是:首先將純化后的蛋白質用消化酶(如胰酶)水解成肽段,肽段混合物經過除鹽后首先進入液相色譜,實現初步分離,以降低樣品復雜度。經液相色譜分離后的肽段先經過質譜的第一級--離子源(ESI,MALDI)使肽段離子化并形成分散的離子。接著選擇部分強度較高的肽段母離子進入質譜的第二級系統(碎裂源)做進一步碎裂,使肽段離子被打碎成更小的、有規律的碎片離子。理論上蛋白質序列經過酶解后形成肽段,肽段再經過理論二級碎裂形成理論的二級碎片離子譜圖。然后用實驗產生的MS/MS譜圖信息(主要包括質荷比、離子強度)與理論產生的MS/MS譜圖信息比對,通過比較兩者的相似度來確定譜圖對應哪個肽段/蛋白(如圖1)。其中理論蛋白質數據庫來自基因組預測或前期實驗驗證等數據。
目前,常用的數據庫有NCBI-nr( http://www.ncbi.nlm.nih.gov/) 、Uniprot( http://www.uniprot.org/)等。常用的搜索引擎有Mascot( http://www.matrixscience.com/) 、Proteinpilot
(http://www.absciex.com/products/software/proteinpilot-software)。Mascot一度被業界奉為鑒定軟件的金標準,但其算法更偏向于鑒定長度較短的肽段序列。而Proteinpilot由于采用自動容錯匹配、對所有可能的蛋白修飾采用了盲搜的策略,無肽段長度偏向性,鑒定效果更好。

圖1 蛋白質組鑒定流程圖
1、通量大:一次實驗可鑒定6000多種蛋白質(以人HepG2為例);
2、結果準確可靠:質譜儀的質量精度可達到1ppm以下,分辨率能達到4x104以上,可提供高質量的數據。
3、適用范圍廣泛:對物種要求無限制,理論上適用于任何物種。
經典案例:
題目:A draft map of the human proteome(人類蛋白質組草圖繪制)
期刊:Nature
主要技術:LC-MS/MS
文章摘要:采用LC-MS/MS技術,對30個來自正常人類的不同組織、器官(17個成年人組織、7個胎兒組織、6個初級純化造血細胞)樣品進行了深度蛋白質組解析,共鑒定到17294個蛋白,占人類全部有注釋編碼蛋白基因的84%。本文同時采用蛋白質組和基因組學(Proteogenomic)的方法發現了一些新的編碼區(這些區域過去被認為是非編碼區)。這一海量蛋白質組數據將會補充現有的基因組和轉錄組數據,從而加速健康、疾病方面的生物醫學研究。
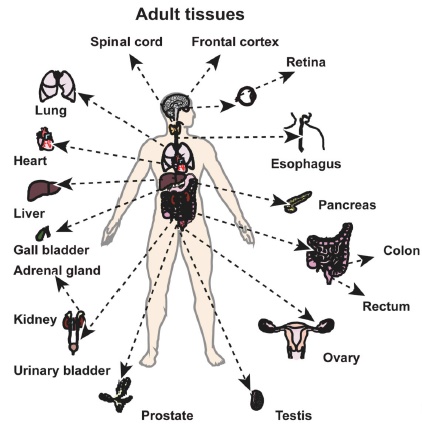
圖2 人體中不同樣品來源
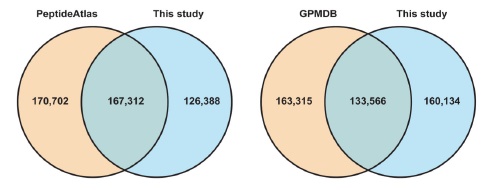
圖3 鑒定結果與數據庫PeptideAtlas 和GPMDB收錄的結果比較



